
INFERENCE DE LA STRUCTURE GENETIQUE D’UNE … · génétique des populations en ce sens qu’il...
22
INFERENCE DE LA STRUCTURE GENETIQUE D’UNE POPULATION TEST DU LOGICIEL STRUCTURE (VERSION 2.3.3) ET APPROCHE DES MODELES MIS EN ŒUVRE PAR LE LOGICIEL LAYLA EL ASRI ENSIMAG 2EME ANNEE – MMIS BIOINFO TRAVAUX D’ETUDES ET DE RECHERCHE TUTEURS : OLIVIER FRANÇOIS ET FLORENCE MARANINCHI
Transcript of INFERENCE DE LA STRUCTURE GENETIQUE D’UNE … · génétique des populations en ce sens qu’il...

INFERENCE DE LA STRUCTURE
GENETIQUE D’UNE POPULATION
TEST DU LOGICIEL STRUCTURE (VERSION
2.3.3) ET APPROCHE DES MODELES MIS EN
ŒUVRE PAR LE LOGICIEL
L A Y L A E L A S R I E N S I M A G
2 E M E A N N E E – M M I S B I O I N F O T R A V A U X D ’ E T U D E S E T D E R E C H E R C H E
T U T E U R S : O L I V I E R F R A N Ç O I S E T F L O R E N C E M A R A N I N C H I

2
S O M M A I R E
1. La génétique des populations
2. Etudes théoriques de différents
modèles
3. Le logiciel MS
4. Le logiciel STRUCTURE
5. Expériences
6. Analyse des résultats et conclusion
7. Bibliographie

3
P R E A M BU L E
Notre travail a été effectué au laboratoire TIMC -Imag sous la direction d ’Oliver François et a concerné l ’étude d ’une méthode de clustering implémentée par le logiciel STRUCTURE. Lorsque l ’on s’ intéresse à l ’évolutio n du patrimoine génétique d’une espèce , la population est souvent prise comme unité de base et i l est uti le de pouvoir répar tir les individus d’un échantil lon au sein de populations . Plusieurs approches sont alors envisageables : on peut chercher à grouper les individus d’un échantil lon en différentes populations ou chercher, à par t ir de la connaissance d’un cer tain nombre de populations, la provenance d’un individu d’orig ine inconnue.
Nous nous intéresserons ici à la première approche et à la méthode dite de « clustering » ayant pour but de grouper des individus en populations sur la base de leur génotype. Un logiciel , largement uti l isé en génétique des populations, se propose d’implémenter cette méthode, le logicie l STRUCTURE.
Notre travail consistera en l ’étude de ce logiciel et des algorithmes uti l isés par celui -c i af in de déterminer la por tée et les l imitat ions de ceux-ci . Pour cela, nous procèderons de la sor te : nous uti l iserons un logiciel permettant de générer des jeux de données génétiques à par tir de modèles prédéfinis, le logiciel MS. Nous ferons trai ter les données générées par ce logiciel par STRUCTURE puis nous comparerons le résultat obtenu avec celui attendu. Nous commencerons par présenter les différentes notions de génétique des populations qui seront uti l isées au cours de nos travaux futurs avant de présenter les expériences réalisées avec les logiciels MS et STRUCTURE avant de conclure quant à l ’efficacité de la méthode de « clustering » implémentée par le logiciel STRUCTURE.
Population : ensemble d'individus d'une même espèce vivante se perpétuant dans un territoire donné

4
L A G E N E T I QU E D E S P O P U L AT I O N S
OBJET DE LA GENETIQUE DES POPULATIONS
La génétique des populations traite de l’impact des différentes forces évolutives (sélection, mutations, migration) sur la répartition de la diversité génétique entre les populations et dans les populations.
La description de la composition génétique d’une population diploïde se fait via différents indicateurs :
Les fréquences génotypiques : il s’agit des fréquences
des différents génotypes au locus considéré. L’ensemble des fréquence génotypiques donne la structure génotypique de la population pour ce locus.
Les fréquences alléliques (ou géniques) : il s’agit des
fréquences dans la population des différents allèles au locus considéré. Leur connaissance donne la structure allélique (génique) de la population.
L’ensemble formé par la structure génotypique et la structure
allélique d’une population constitue la structure génétique de la population.
Cellule diploïde : les chromosomes qu’elle contient sont présents par paires.
Locus : emplacement physique précis et invariable sur un chromosome. Génotype : ensemble ou partie donnée de la composition génétique d’un individu. Allèles d’un gène : les allèles d’un gène correspondent aux différentes versions de ce gène (leurs séquences de nucléotides diffèrent).

5
DESCRIPTION FORMELLE DE LA STRUCTURE GENOTYPIQUE ET DE LA STRUCTURE ALLELIQUE D’UNE
POPULATION DIPLOIDE
Considérons un gène A en un locus pour une population diploïde.
Structure génotypique en ce locus:
( )
Où ( ) représente la fréquence des individus ayant le génotype AA, est le nombre d’individus ayant ce génotype, et N le nombre total d’individus.
Considérons désormais un gène A possédant de multiples allèles A1, …, An en un
locus pour une population diploïde.
Structure allélique en ce locus :
∑
Où représente la fréquence de l’allèle Ai en le locus considéré.

6
E T U D E S T H E O R I QU E S D E D I F F E R E N T S M O D E L E S
LE PRINCIPE DE HARDY-WEINBERG
Il s’agit ici d’étudier la génétique d’une population dans un cas « idéal », c’est-à-dire, sous les hypothèses suivantes : 1) La panmixie : les gamètes s’associent au hasard par rapport aux gènes considérés. 2) La population est considérée être de taille infinie, ainsi, la fréquence d’un évènement est proche de la probabilité que cet évènement se produise. 3) On considère comme nul l’apport des pressions évolutives suivantes :
Mutation
Sélection
Migration Par ailleurs, la population étudiée est une population diploïde où coexistent deux allèles en un locus : A et a en fréquences respectives p et q = 1 –p. Sous les hypothèses 1), 2), et 3), nous pouvons alors déterminer exactement, la structure génétique de la génération diploïde suivante :
( )
( ) ( )
(par l’hypothèse 2))
( è ) )) En effectuant des calculs similaires pour les autres génotypes, on obtient les résultats suivants :
Gamète Gamètes mâles Ga Gamètes femelles
A
a
A
p2
pq
a
pq
q2
Structure génotypique de la génération diploïde suivante

7
En appliquant l’hypothèse 3), on s’aperçoit que les fréquences alléliques de la nouvelle génération seront respectivement p et q pour les allèles A et a. Ainsi, dans un cas idéal où il y a panmixie pour une population de taille infinie et qui n’est soumise à aucune pression évolutive, les fréquences des gènes et des génotypes ne varient pas d’une génération à l’autre, ce qui constitue le principe de Hardy-Weinberg. Il s’agit d’un état d’équilibre pour la population. L’équilibre de Hardy-Weinberg est un des principes fondamentaux de la génétique des populations en ce sens qu’il s’agit d’un bon descripteur de la structure génétique des populations naturelles, l’hypothèse de panmixie étant souvent respectée alors que les effets des mutations, migrations et sélections ne sont pas suffisamment importants pour faire diverger les fréquences génotypiques étudiées. Nous verrons par la suite que les résultats produits par STRUCTURE incluent dans leurs hypothèses le fait que les populations étudiées soient à l’équilibre de Hardy-Weinberg.
LE MODELE DE WRIGHT-FISHER
Le modèle de Wright-Fisher permet de représenter l’évolution d’un allèle à l’intérieur d’une population diploïde ou haploïde. Il sera notamment utilisé par la suite dans le modèle du coalescent. Nous considèrerons le modèle suivant :
La population considérée est de taille constante. On supposera une taille de N individus diploïdes soit 2N individus haploïdes.
Les générations sont disjointes c’est-à-dire que tous les individus de la génération précédente décèdent et donnent naissance à tous les individus de la génération suivante.
Il n’y a aucune sélection.
Les accouplements se font de façon aléatoire (population panmictique). 1. Principe Sous le modèle de Wright-Fisher, l’algorithme permettant de retracer l’évolution d’un allèle est le suivant :
Au temps 0, la population est constituée de N individus possédant chacun deux copies de l’allèle auquel on s’intéresse.
Les N individus de la génération 1 sont générés indépendamment les uns des autres de la façon suivante :
1) Un premier parent est choisi de manière équiprobable dans la génération 0.
Cellule haploïde : Les chromosomes qu’elle contient ne sont présents qu’en un seul exemplaire.

8
2) On choisit, de manière équiprobable, un allèle de ce premier parent et l’on en place une copie comme premier allèle de l’individu de la génération 1.
3) De la même manière, on choisit le second parent de façon équiprobable dans la génération 0.
4) Enfin, on choisit de manière équiprobable un allèle du second parent et l’on en place une copie comme second allèle de l’individu de la génération 1.
Les générations suivantes sont alors générées de la même façon.
.
Figure 1 Modèle de Wright-Fisher pour une population diploïde
2. Etude probabiliste Nous déduisons qu’à l’issue de cette simulation, le nombre de descendants dans la génération t + 1 de l’allèle k de la génération t suit une loi binomiale de paramètres
( 2N,
) * 2 2 +. En effet, la probabilité qu’à la génération t + 1, il y
ait i représentants de l’allèle k de la génération t est égale au nombre de « succès » de l’évènement : « 1 allèle de la génération t + 1 descend de l’allèle k de la génération t »
et la probabilité de cet évènement est égale à
puisque les allèles sont tirés de
façon équiprobable. Ce modèle ainsi défini va nous permettre désormais d’introduire la théorie du coalescent.

9
LE MODELE DU COALESCENT
Problématique : Avant l’introduction de la théorie du n-coalescent, les modèles utilisés en génétique des populations représentaient l’évolution d’une population dans le sens naturel du temps : on déduisait, à partir d’une population de départ, des résultats concernant les générations subséquentes engendrées selon des règles bien précises. L’étude des polymorphismes se faisait ainsi : une fois les données collectées, on émettait un scénario possible de généalogie de l’échantillon en estimant l’arbre généalogique sans prendre en compte les possibles recombinaisons. 1. Principe L’approche de la génétique des populations par le modèle du n-coalescent est différente, il s’agit d’une approche rétrospective. En effet, on travaille dans ce cas en examinant l’histoire ancestrale d’un échantillon de départ. L’étude des polymorphismes se fait alors ainsi : une fois les données collectées et toutes les généalogies possibles prises en compte, y compris celles incluant des recombinaisons génétiques, on estime l’arbre généalogique de l’échantillon grâce à des méthodes statistiques de maximisation de la vraisemblance des données. Les avantages de cette méthode sont que celle-ci permet de choisir entre les différents modèles possibles en utilisant des méthodes statistiques standards et qu’elle permet de prendre en compte aisément différents phénomènes géographiques tels que les migrations et les ad mixture. 2. Temps de coalescence Pour nos travaux futurs, il nous sera utile de pouvoir estimer le temps écoulé entre l’apparition d’une mutation et celle d’un allèle dans une population. La probabilité que deux lignées coalescent directement à la génération précédente correspond à la probabilité que ces deux lignées possèdent un parent commun. Considérons une population diploïde de taille N, pour chaque allèle, il y a 2N parents possibles donc la
probabilité que deux allèles possèdent un parent commun est égale à
et inversement, la probabilité que les deux lignées ne coalescent pas à la
génération précédente vaut
Ainsi, si l’on considère T la variable
aléatoire correspondant au temps de coalescence entre deux lignées, T
est une variable discrète suivant une loi géométrique de paramètre
, la
probabilité que deux lignées coalescent au temps t correspond à la probabilité qu’elles ne coalescent pas au t – 1 précédentes générations
pour coalescer à la t-ième génération, soit, en notant ( ) cette
probabilité, on a : ( ) (
)
. En outre, dans le
cas qui nous intéresse, nous étudions des populations de grandes tailles, nous pouvons alors approcher cette distribution par une distribution
exponentielle, soit : ( ) (
)
.
Polymorphisme génétique : coexistence dans une population de plusieurs allèles pour un gène ou un locus donné
Recombinaison génétique : le phénomène conduisant à l’apparition, dans une cellule ou dans un individu, de gènes ou de caractères héréditaires dans une association différente de celle observée chez les cellules ou individus parentaux.

10
Figure 2 Exemple d'arbre de coalescence généré avec MS pour une population haploïde de 6 individus.
EFFECTIF EFFICACE D’UNE POPULATION
1. Définition Dans les populations naturelles, tous les individus ne participent pas au processus reproductif ce qui permet de définir un effectif « efficace » de la population comme étant l’effectif d’une population idéale (de type Wright-Fisher) pour laquelle on aurait une fluctuation du polymorphisme équivalente à celle de la population naturelle. Il s’agit
donc du nombre d'individus d'une population idéale pour lequel on aurait un degré de dérive génétique équivalent à celui de la population réelle. Pour une population, l’effectif efficace est alors défini comme la moyenne harmonique des effectifs précédents de la population :
∑
2. Cas de l’espèce humaine Dans le cas de l’espèce humaine, l’hypothèse courante est que la population était constante à 10 000 individus d’il y a 200 000 ans à il y a 100 000 ans puis, a connu une croissance exponentielle, passant de 10 000 à 10 milliards d’individus d’il y a 100 000 ans à aujourd’hui. En comptant une génération tous les 20 ans, on arrive à une moyenne harmonique de 18 648 individus pour les derniers 200 millénaires. Ainsi, on s'attend à ce que la variabilité génétique et moléculaire de l'espèce humaine soit comparable à celle d'une population idéale d'environ 20 000 individus.
7 8 1 2 3 5 4 6
Dérive génétique : modification de la fréquence d'un allèle, ou d'un génotype, au sein d'une population, pour une raison autre que les mutations, la sélection naturelle ou les migrations.

11
L E L O G I C I E L M S Le logiciel MS permet de générer des échantillons de données génétiques sous des hypothèses de migration, mutation, recombinaison… L’algorithme de formation des échantillons est basée sur la théorie du coalescent précédemment décrite.
OBJECTIFS
L’étude du polymorphisme génétique est une question centrale de la génétique des populations : sa compréhension permet de retracer l’histoire d’une population et de ses individus. Le logiciel MS permet d’analyser les différents polymorphismes observés au sein de populations. Pour ce faire, le logiciel propose différents types de simulations de jeux de données en appliquant différentes hypothèses concernant la migration, la recombinaison, etc.
PRINCIPE/LE COALESCENT
Les échantillons sont générés en s’appuyant sur la théorie du coalescent où la généalogie aléatoire de l’échantillon est générée en premier lieu puis les mutations y sont placées de manière aléatoire.
LES PRINCIPAUX PARAMETRES DES SIMULATIONS
Chaque simulation nécessite le réglage de deux paramètres obligatoires : le nombre d’individus par échantillons nsam suivi du nombre d’échantillons nreps, auxquels s’ajoute un troisième paramètre qui dans notre cas sera le paramètre de
mutation . Le paramètre de mutation est défini de la manière suivante : il est égal à
où représente l’effectif efficace de la population diploïde dont les
individus sont issus et le taux de mutation pour le locus considéré. Par exemple, dans le cas de l’étude d’une population humaine, en ce qui
concerne le taux de mutation, il est estimé qu’il se produit environ 2 mutations par base par génération. Ainsi, sur une génération, une base de
nucléotides subira 2 mutations. Ainsi, sera estimé comme étant égal à
2 , étant le nombre de paires de base pour le locus considéré.

12
MODELES
MS permet de simuler différents scénarii d’évolution d’une population. Nous allons présenter ici ceux qui nous ont été utiles au cours de nos expériences. L’ad mixture/Les modèles en île
On modélise ici le scénario suivant : des individus de plusieurs sous-populations migrent de l’une à l’autre. Les paramètres à régler sont :
Le nombre d’individus ainsi que leur répartition au sein des différentes sous-populations
Le taux de mutation
Le taux de migation pour chaque sous-population (c’est-à-dire, pour chaque sous-population, le nombre de migrants par génération)
Les microsatellites Chez les eucaryotes, les microsatellites sont généralement présents en grand
nombre dans le génome d’une espèce. Un microsatellite est une séquence d’ADN constituée d’une répétition continue de motifs composés de 2 à 10 nucléotides. Les motifs répétés sont souvent des motifs de 1, 2, 3, ou 4 paires de base. Par exemple, le motif composé de deux nucléotides (CA) est présent dans l’ADN tous les 25 kilo bases.
Le paquetage fournit par l’auteur de MS contient la fonction microsat.c qui permet de transposer le format de sortie des données issues d’une simulation en terme de variation du nombre de microsatellites pour chaque individu. L’algorithme implémenté par MS est une marche aléatoire : la variable aléatoire correspondant au nombre de microsatellites est incrémentée de 1 ou diminuée de 1 d’une génération à l’autre de façon équiprobable. L’intérêt de l’utilisation de ce paquetage est qu’il fournit des données qui nécessitent moins de manipulations avant d’être passées à STRUCTURE.

13
L E L O G I C I E L S T RU C T U R E
OBJECTIFS
Le programme STRUCTURE implémente une méthode de clustering pour l’inférence de la structure génétique de populations à partir de données génétiques sous la forme de marqueurs indépendants. L’objectif est de , sur la base de leur génotype, grouper les individus en populations mais aussi estimer leur degré d’introgression/hybridation, inférer l’origine d’un locus en particulier et estimer le nombre de populations.
HYPOTHESES
Toutes les populations inférées sont considérées être à l’équilibre de Hardy-Weinberg et on fait l’hypothèse d’équilibre de liaison entre loci au sein des populations. La notion d’équilibre de liaison génétique entre deux loci reflète le fait que les allèles présents à l’un des locus s’associent au hasard aux allèles présents à l’autre locus. Ainsi, la fréquence d’observation au sein d’une population d’une combinaison d’allèles est égale au produit des fréquences d’observations individuelles des allèles.
PRINCIPE/L’INFERENCE BAYESIENNE
1) Inférence de la structure génétique L’algorithme utilisé est une algorithme d’inférence reposant sur des
principes d’analyse statistique bayésienne.
Le principe est le suivant : Soient X le vecteur aléatoire correspondant aux génotypes des individus échantillonnés, Z correspondant aux populations d’origine des individus, et P aux fréquences alléliques dans chacune des populations.
Sous les hypothèses précédemment citées, la loi de est complètement connue. Alors, après avoir spécifié les priors Z et P, la règle de Bayes permet
de déduire la loi de (en effet,
( ) ( ) ( ) ( )). Pour ce faire, un algorithme basé sur la méthode de Monte Carlo permet d’obtenir des couples de solutions approchées (Z1, P1), (Z2, P2)…
Liaison génétique : il s’agit de la tendance, pour des allèles de différents gènes, d’être transmis ensemble d’une génération à l’autre. Haplotype : un haplotype désigne, chez un individu, l’ensemble des différents gènes présents et génétiquement liés sur un même chromosome.

14
2) Inférence du nombre K de populations Selon la règle de Bayes, la détermination du nombre K de populations se fait à
posteriori, en sachant : ( ) ( ) ( ). Cependant, la distribution ainsi définie à posteriori peut varier de manière notable en fonction des hypothèses de
modélisation et l’estimation de ( ) est une tâche délicate. Ainsi, une méthode
d’estimation ad hoc a été préférée, supposant que suit une loi exponentielle.
MODELES
Le logiciel STRUCTURE implémente quatre modèles de base pour l’ascendance d’un individu : le modèle sans introgression (no ad mixture model), le modèle avec introgression (ad mixture model), le modèle chromosomique avec recombinaison (linkage model) et le modèle avec dérive (using prior population information). Notons K le nombre de populations inférées.
Le modèle sans introgression : On émet dans ce modèle l’hypothèse selon laquelle chaque individu provient
exclusivement d’une des K populations inférées c’est-à-dire que tous les gènes d’un individu proviennent d’une unique population parmi les K possibles. Le résultat sera alors la probabilité a posteriori que l’individu i appartienne à la population k. Le
prior, ici la probabilité qu’un individu i appartienne à la population k est égal à
.
Le modèle avec introgression :
Dans ce modèle, les différents gènes d’un individu peuvent être hérités de différentes populations. On s’intéresse alors à la proportion Q des gènes d’un individu provenant de la population k. Le résultat délivré par STRUCTURE sera alors l’estimation moyenne a posteriori de ces différentes proportions.
Le modèle chromosomique avec recombinaison : Ce modèle permet de prendre en compte le déséquilibre de liaison lié à
l’introgression c’est-à-dire les corrélations entre loci génétiquement liés du fait d’un évènement d’ad mixture récent entre les populations. Le modèle de base est le suivant : si l’on remonte de t générations dans le passé, il y a eu un évènement d’ad mixture qui a mélangé les K populations. Un chromosome d’un individu descendant de ce mélange sera alors constitué de différentes « portions » héritées des différentes populations. Le déséquilibre de liaison apparait alors parce que des allèles génétiquement liés sont souvent sur la même « portion » de chromosome et proviennent donc d’un même ancêtre.
Le modèle avec dérive : Ce modèle prend en compte des informations autres que les données purement
génétiques telles que des caractéristiques physiques des individus échantillonnés ou des répartitions géographiques.

15
E X P E R I E N C E S
PROCEDE
Nous allons partir de modèles créés grâce au logiciel ms que nous allons analyser avec le logiciel Structure afin de tester le bon fonctionnement de celui-ci et de déterminer ses limitations.
Première expérience
« LE MODELE EN ILES »
Présentation du modèle :
Il s’agit ici d’étudier un modèle avec deux sous-populations qui reçoivent chacune des migrants venant de l’autre sous-population. Objectifs :
L’intérêt est de s’assurer que STRUCTURE détecte bien la présence des deux sous-populations à partir du modèle généré par MS. Pour cela, nous allons faire varier la composition des populations et les taux de migrations afin d’observer le comportement de STRUCTURE face aux données reçues. Création des données avec ms :
Nous allons considérer un locus humain de 1000 paires de bases. Nous avons
donc : Nous nous intéresserons à 10 000 individus répartis en deux sous-populations. Nous allons commencer notre étude en faisant varier le taux de migration que nous choisirons symétrique dans un premier temps.

16
Nous avons donc effectué un premier test en simulant un modèle en îles de la manière suivante :
Au départ, 10 000 individus étaient répartis en deux sous-populations distinctes (chacune comptant 5 000 individus). Nous avons alors défini la matrice de migration M suivante :
(
)
Ainsi, à chaque génération et pour chacune des deux populations, 3 individus
sur 100 proviennent de l’autre sous-population. La taille des deux sous-populations étant constante, à chaque génération 150 individus d’une des sous-populations proviennent de l’autre sous-population.
Par ailleurs, nous avons choisi d’étudier ces populations à partir des données concernant 50 loci, nombre qui au vu des manipulations préalables qui nous avait permises de prendre en main STRUCTURE nous paraissait suffisant. Nous avons donc simulé ces données via MS à l’aide de la commande suivante :
./ms 10000 50 -t 1 -I 2 5000 5000 -ma 0.0 0.03 0.03 0.0
Les données récoltées ont ensuite été manipulées à l’aide du logiciel R afin d’avoir un jeu de données exploitables par STRUCTURE.
Chaque génération, 0.03 % de la pop. 1 provient de la pop. 2
Chaque génération, 0.03 % de la pop. 1 provient de la pop. 2
Sous-population 1 Sous-population 2
17
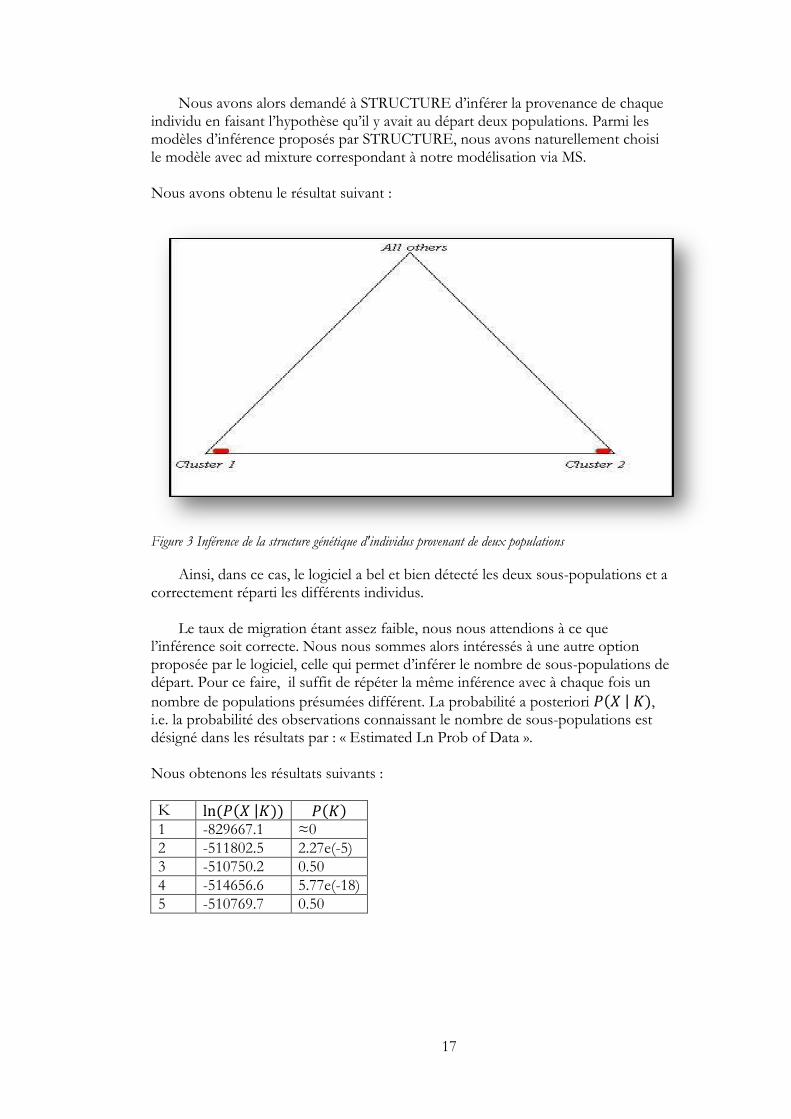
Nous avons alors demandé à STRUCTURE d’inférer la provenance de chaque individu en faisant l’hypothèse qu’il y avait au départ deux populations. Parmi les modèles d’inférence proposés par STRUCTURE, nous avons naturellement choisi le modèle avec ad mixture correspondant à notre modélisation via MS. Nous avons obtenu le résultat suivant :
Figure 3 Inférence de la structure génétique d'individus provenant de deux populations
Ainsi, dans ce cas, le logiciel a bel et bien détecté les deux sous-populations et a correctement réparti les différents individus.
Le taux de migration étant assez faible, nous nous attendions à ce que l’inférence soit correcte. Nous nous sommes alors intéressés à une autre option proposée par le logiciel, celle qui permet d’inférer le nombre de sous-populations de départ. Pour ce faire, il suffit de répéter la même inférence avec à chaque fois un
nombre de populations présumées différent. La probabilité a posteriori ( ), i.e. la probabilité des observations connaissant le nombre de sous-populations est désigné dans les résultats par : « Estimated Ln Prob of Data ». Nous obtenons les résultats suivants :
K l ( ( )) ( ) 1 -829667.1 ≈0
2 -511802.5 2.27e(-5)
3 -510750.2 0.50
4 -514656.6 5.77e(-18)
5 -510769.7 0.50

18
Associés aux répartitions suivantes :
Nombre de populations K = 2 K = 3
K = 4 K = 5
Il est ainsi surprenant de constater que d’après l’inférence réalisée par
STRUCTURE, le nombre de sous populations le plus vraisemblable pour ce génotype est de 3. Par ailleurs, pour ce nombre, STRUCTURE estime que 50 % des individus proviennent de deux sous-populations et que les 50 % restants proviennent d’une troisième sous-population. Ainsi, dans ce cas, le logiciel ne trouve pas le bon résultat.
En revanche, lorsqu’on lui fournit le nombre correct de sous-populations de
départ, STRUCTURE donne la bonne inférence en répartissant les individus en deux sous-populations distinctes et ce, même pour des taux de migration élevés.

19
Deuxième expérience
« LES MICROSATELLITES »
Après avoir testé le cas simple de deux populations qui échangent des migrants,
nous avons voulu testé STRUCTURE sur un cas plus compliqué : le cas où différentes populations se mélangent à des temps donnés. STRUCTURE fournit alors différentes inférences et indique la vraisemblance de chacune. Nous allons donc lancer plusieurs inférences et analyser les résultats produits par STRUCTURE.
Nous avons pour cela utilisé la fonction microsat fournie par MS à l’aide de laquelle nous avons simulé un jeu de données concernant 377 loci chez 98 individus et après traitement des données via R, nous avons lancé l’analyse des données sous STRUCTURE en faisant l’hypothèse qu’il y avait au départ 4 populations. Le scénario est le suivant : au départ, il y avait 4 populations distinctes. Au bout d’un certain temps t1, deux sous-populations se sont mélangées et au bout d’un certain temps t2, une troisième s’est mélangé à la population résultante. Ensuite, au bout d’un temps t3, la dernière sous-population s’est mélangée à la population formée des trois autres sous-populations.
On a représenté ici le scénario
simulé sous MS où les 4 sous-populations se mélangent
progressivement au cours du temps.
Nous avons simulé les données à l’aide de la commande MS suivante : ./ms.exe 98 377 –t 7.1 -I 4 30 30 24 14 –ej 0.05 2 1 –ej .0.15 3
1 –ej 0.6 4 1 –n 1 6 –n 2 7 –n 3 700 –n 4 11 –g 1 10 –g 2 10 –g 3
18 –g 4 14 –em 0.12 1 3 200 | ./microsat
Les options utilisées ici précisent les temps t1, t2, et t3, ainsi que des hypothèses sur les tailles des populations et leur croissance au cours du temps. Nous avons ensuite demandé l’analyse des données via STRUCTURE cinq fois en gardant les mêmes paramètres, nous avons obtenu les résultats suivants :
t = 0
t3
t2 t1
1 2 3 4
t

20
Simulation n°1 Simulation n°2 Simulation n°3
Simulation n°4 Simulation n°5 Les vraisemblances des différentes simulations sont les suivantes :
Numéro de la simulation l ( ( )) 1 -69731.1
2 -67103.7
3 -66912.5
4 -66141.8
5 -66170.4
Ainsi, au vu des données fournies à STRUCTURE, le modèle le plus vraisemblable est celui de la simulation 4 alors que le modèle le plus proche du scénario original est les modèle de la simulation n°5. Par ailleurs, on peut s’étonner ici du fait d’obtenir avec les mêmes paramètres des résultats très différents.

21
ANALYSE DES RESULTATS ET CONCLUSION
INFERENCE DU NOMBRE DE POPULATIONS DE DEPART
L’expérience ici décrite ainsi que celles que l’ont peut trouver dans la littérature montrent que l’estimation a posteriori du nombre de populations est une tâche délicate et imprécise et les auteurs du programme précisent bien dans la documentation que cette estimation n’est qu’une approximation ad hoc du nombre de populations.
Ainsi, il ne vaut mieux pas utiliser STRUCTURE pour inférer le nombre de populations de départ d’un échantillon. En effet, en augmentant K, on augmente le degré de liberté du modèle donc on s’attendrait théoriquement à une augmentation de la vraisemblance des données or comme on a pu le constater, ce n’est pas le cas. Ainsi, il y a un problème de convergence dans le modèle d’estimation a posteriori de K comme étant celui qui maximise la vraisemblance des données. ETUDE DE MODELES COMPLEXES AVEC DE MULTIPLES EVENEMENTS DE COALESCENCES Notre seconde expérience montre des comportements différents du logiciel sans modification du jeu de données ni des paramètres de simulation. Sur cinq simulation, une seule était correcte et ce n’était pas celle qui était jugée comme étant la plus vraisemblable par STRUCTURE. CONCLUSION Pour rappel, l’algorithme de clustering utilisé par STRUCTURE est le suivant : En notant X les génotypes des individus échantillonnés, Z les populations d'origine, P les fréquences alléliques dans les populations et Q proportion des locus provenant de la population K : Initialisation :
1. Les valeurs de Z sont tirées dans le prior Boucle :
2. Les fréquences P sont estimées à partir des individus assignés à chaque population lors de l’étape précédente
3. Les individus sont assignés aux populations Z à partir des fréquences alléliques P calculées précédemment
Des paramètres de simulation adaptés (i.e. un nombre élevé de tours de boucle) devraient permettre à P et K de converger vers les bonnes distributions. Cependant, nous avons vu à travers les deux cas précédents que ces deux variables ne convergent pas toujours vers les bonnes distributions, et ce probablement à cause des estimations approchées des couples (Z1, P1), (Z2, P2)… Des améliorations futures pourraient donc être apportées en explorant d’autres méthodes plus stables pour l’estimation de ces couples de valeurs.

22
BIBLIOGRAPHIE
Précis de génétique des populations, Jean-Pierre Henry, Pierre-Henri Gouyon, Masson, Paris 1998 A primer of population genetics, Daniel L.Hartl, sinauer 3e éd., 2000. ISBN 0-87893-304-2. Genealogical trees, coalescent theory and the analysis of genetic polymorphisms, NA Rosenberg, M Nordborg - Nature reviews genetics, 2002
Hudson, R.R. (2002) Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18: 337-338.
, Inference of population structure using multilocus genotype data Jonathan K. Pritchard, Matthew Stephens, and Peter Donnelly, Department of statistics, University of Oxford


















