
HANAC Syndrome Col4a1 Mutation Causes Neonate...
13
BASIC RESEARCH www.jasn.org HANAC Syndrome Col4a1 Mutation Causes Neonate Glomerular Hyperpermeability and Adult Glomerulocystic Kidney Disease Zhiyong Chen,* Tiffany Migeon,* † Marie-Christine Verpont,* † Mohamad Zaidan,* Yoshikazu Sado, ‡ Dontscho Kerjaschki, § Pierre Ronco,* †| and Emmanuelle Plaisier* †| *Institut National de la Santé et de la Recherche Médicale, Unité Mixte de Recherche (UMR)S 1155, Paris, France; † Sorbonne University, Université Pierre et Marie Curie, Paris 06, UMR_S 1155, Paris, France; ‡ Division of Immunology, Shigei Medical Research Institute, Okayama, Japan; § Clinical Institute of Pathology, Medical University of Vienna, Vienna, Austria; and | Assistance Publique-Hôpitaux de Paris, Department of Nephrology and Dialysis, Tenon Hospital, Paris, France ABSTRACT Hereditary angiopathy, nephropathy, aneurysms, and muscle cramps (HANAC) syndrome is an autosomal dominant syndrome caused by mutations in COL4A1 that encodes the a1 chain of collagen IV, a major component of basement membranes. Patients present with cerebral small vessel disease, retinal tortuosity, muscle cramps, and kidney disease consisting of multiple renal cysts, chronic kidney failure, and sometimes hematuria. Mutations producing HANAC syndrome localize within the integrin binding site containing CB3[IV] fragment of the COL4A1 protein. To investigate the pathophysiology of HANAC syndrome, we generated mice harboring the Col4a1 p.Gly498Val mutation identi fied in a family with the syndrome. Col4a1 G498V mutation resulted in delayed glo- merulogenesis and podocyte differentiation without reduction of nephron number, causing albuminuria and he- maturia in newborns. The glomerular defects resolved within the first month, but glomerular cysts developed in 3-month-old mutant mice. Abnormal structure of Bowman’s capsule was associated with metalloproteinase in- duction and activation of the glomerular parietal epithelial cells that abnormally expressed CD44, a-SMA, ILK, and DDR1. Inflammatory infiltrates were observed around glomeruli and arterioles. Homozygous Col4a1 G498V mu- tant mice additionally showed dysmorphic papillae and urinary concentration defects. These results reveal a de- velopmental role for the a1a1a2 collagen IV molecule in the embryonic glomerular basement membrane, affecting podocyte differentiation. The observed association between molecular alteration of the collagenous network in Bowman’s capsule of the mature kidney and activation of parietal epithelial cells, matrix remodeling, and inflam- mation may account for glomerular cyst development and CKD in patients with COL4A1-related disorders. J Am Soc Nephrol 27: 1042–1054, 2016. doi: 10.1681/ASN.2014121217 Type IV collagen is a major component of basement membranes (BMs). It consists of six homologous a chains designated a1(IV) –a6(IV) encoded by COL4A1–COL4A6 genes. 1 Each a-chain contains a large collagenous domain composed of Gly-Xaa-Yaa repeats, a C-terminal noncollagenous region, and a small N-terminal domain. Three a-chains assemble to form three distinct triple-helical molecules. 2 The a1a1a2(IV) trimer is ubiquitously expressed in em- bryonic BMs, including embryonic glomerular base- ment membranes (GBMs), and persists in most adult organs. In the mature kidney, it represents the main collagen IV molecule of the tubular, vascular, and Bowman’ s capsule BM, whereas it remains only weakly expressed in the GBM. 3 The a3a4a5(IV) and a5a5a6(IV) trimers display a more restricted Received December 12, 2014. Accepted June 16, 2015. Published online ahead of print. Publication date available at www.jasn.org. Correspondence: Prof. Emmanuelle Plaisier, Department of Nephrology and Dialysis, INSERM UMR_S 1155, Hôpital Tenon, 4 rue de la Chine, 75020 Paris, France. Email: emmanuelle.plaisier@ tnn.aphp.fr Copyright © 2016 by the American Society of Nephrology 1042 ISSN : 1046-6673/2704-1042 J Am Soc Nephrol 27: 1042–1054, 2016
Transcript of HANAC Syndrome Col4a1 Mutation Causes Neonate...

BASIC RESEARCH www.jasn.org
HANAC Syndrome Col4a1 Mutation Causes NeonateGlomerular Hyperpermeability and AdultGlomerulocystic Kidney Disease
Zhiyong Chen,* Tiffany Migeon,*† Marie-Christine Verpont,*† Mohamad Zaidan,*Yoshikazu Sado,‡ Dontscho Kerjaschki,§ Pierre Ronco,*†| and Emmanuelle Plaisier*†|
*Institut National de la Santé et de la Recherche Médicale, Unité Mixte de Recherche (UMR)S 1155, Paris, France;†Sorbonne University, Université Pierre et Marie Curie, Paris 06, UMR_S 1155, Paris, France; ‡Division of Immunology,Shigei Medical Research Institute, Okayama, Japan; §Clinical Institute of Pathology, Medical University of Vienna,Vienna, Austria; and |Assistance Publique-Hôpitaux de Paris, Department of Nephrology and Dialysis, Tenon Hospital,Paris, France
ABSTRACTHereditary angiopathy, nephropathy, aneurysms, and muscle cramps (HANAC) syndrome is an autosomaldominant syndromecausedbymutations inCOL4A1 that encodes thea1chainof collagen IV, amajor componentof basement membranes. Patients present with cerebral small vessel disease, retinal tortuosity, muscle cramps,and kidney disease consisting of multiple renal cysts, chronic kidney failure, and sometimes hematuria. MutationsproducingHANAC syndrome localizewithin the integrin binding site containingCB3[IV] fragment of theCOL4A1protein. To investigate the pathophysiology of HANAC syndrome, we generated mice harboring the Col4a1
p.Gly498Val mutation identified in a family with the syndrome. Col4a1 G498V mutation resulted in delayed glo-merulogenesis and podocyte differentiation without reduction of nephron number, causing albuminuria and he-maturia in newborns. The glomerular defects resolved within the first month, but glomerular cysts developed in3-month-old mutant mice. Abnormal structure of Bowman’s capsule was associated with metalloproteinase in-duction and activation of the glomerular parietal epithelial cells that abnormally expressedCD44,a-SMA, ILK, andDDR1. Inflammatory infiltrates were observed around glomeruli and arterioles. HomozygousCol4a1G498Vmu-tant mice additionally showed dysmorphic papillae and urinary concentration defects. These results reveal a de-velopmental role for thea1a1a2collagen IVmolecule in theembryonicglomerularbasementmembrane,affectingpodocyte differentiation. The observed association between molecular alteration of the collagenous network inBowman’s capsule of the mature kidney and activation of parietal epithelial cells, matrix remodeling, and inflam-mation may account for glomerular cyst development and CKD in patients with COL4A1-related disorders.
J Am Soc Nephrol 27: 1042–1054, 2016. doi: 10.1681/ASN.2014121217
Type IV collagen is a major component of basementmembranes (BMs). It consists of six homologous achains designated a1(IV)–a6(IV) encoded byCOL4A1–COL4A6 genes.1 Each a-chain contains alarge collagenous domain composed of Gly-Xaa-Yaarepeats, a C-terminal noncollagenous region, and asmall N-terminal domain. Three a-chains assembleto form three distinct triple-helical molecules.2 Thea1a1a2(IV) trimer is ubiquitously expressed in em-bryonic BMs, including embryonic glomerular base-mentmembranes (GBMs), and persists inmost adultorgans. In the mature kidney, it represents the maincollagen IV molecule of the tubular, vascular, and
Bowman’s capsule BM, whereas it remains onlyweakly expressed in the GBM.3 The a3a4a5(IV)and a5a5a6(IV) trimers display a more restricted
Received December 12, 2014. Accepted June 16, 2015.
Published online ahead of print. Publication date available atwww.jasn.org.
Correspondence: Prof. Emmanuelle Plaisier, Department ofNephrology and Dialysis, INSERM UMR_S 1155, Hôpital Tenon, 4rue de la Chine, 75020 Paris, France. Email: [email protected]
Copyright © 2016 by the American Society of Nephrology
1042 ISSN : 1046-6673/2704-1042 J Am Soc Nephrol 27: 1042–1054, 2016

distribution, including in the kidney, wherethe former is expressed in the mature GBMand distal tubule BM, and the latter is selec-tively found in the adult Bowman’s capsule.3
Collagen IV networks play a crucial struc-tural role inBMs, but they are also involved inembryogenesis, cell migration and differenti-ation, angiogenesis, tumor growth, and me-tastasis.These functionsaremediated throughinteraction with cell receptors, mainly in-tegrins and discoidin domain receptors 1(DDR1).4–6
Alport syndrome was the first collagenIV–inherited disease characterized withmutations affecting COL4A3, COL4A4, orCOL4A5 genes.3 COL4A1 mutations weremore recently identified in families affectedby autosomal dominant porencephaly andcerebral small vessel disease.7–11 Eye abnor-malities, such as cataract, anterior segmentdysgenesis, and retinal tortuosity, were occa-sionally reported together with the brain de-fects.9–12 Concomitantly, severalCol4a1 ENUmutant mice were analyzed, showing severebrain disease and eye alterations similar to theones observed in patients.7,13–15
We have characterized a distinct systemicphenotype associated with COL4A1 muta-tions in six families, which we named hered-itary angiopathy, nephropathy, aneurysms,and muscle cramps (HANAC).16 Affectedpatients usually show muscle cramps, mildcerebral small vessel disease, retinal arteriolartortuosity, intracranial aneurysms, and a
Figure 1. Col4a1 G498V mutant newborns show albuminuria and hematuria associ-ated with podocyte and GBM defects at P0. (A) Urine dipstick tests revealed heavyproteinuria and hematuria in heterozygous (+/G498V) and homozygous (G498V/G498V) Col4a1 mutant mice at P0. (B) Phase-contrast microscopy analysis of spoturine sample of Col4a1 homozygous mutant P0 mice showed numerous dimorphicerythrocytes (arrow). (C) Quantification of albuminuria in wild-type (+/+), heterozygousand homozygous mutant newborns at P0. Values represent mean6SEM ***P,0.0001compared with +/+. (D and E) Compared with wild-type animals (D), sagittal sectionsof newborn kidneys in Col4a1 homozygous mutant mice at P0 (E) showed enlargedBowman’s spaces with retracted capillary tufts in a subset of glomeruli (white arrow)and dilated proximal tubular sections with flattened epithelial cells. Note also thewidening of the collecting duct lumen in the medulla (periodic acid–Schiff). (F and G)Semi-thin sections of immature glomeruli after the formation of the first capillary loopsin wild-type (F) and homozygous mutant (G) P0 kidneys showing podocyte disorga-nization in mutant glomeruli (toluidine blue stain). (H and I) Mature glomeruli in thedeep cortex of wild-type (H) and homozygous mutant (I) P0 kidneys. Mutant miceshowed dilated glomerular capillaries (toluidine blue stain). (J) Erythrocytes in dilatedtubule sections with epithelial cell flattening, adjacent to a glomerulus with retractedcapillary tuft and enlarged Bowman’s space in a homozygous mutant kidney (Masson’s
trichrome). (K) Intracytoplasmic eosinophilicvacuoles in proximal tubular cells of homozy-gous mice (hematoxylin and eosin). (L and M)Collectingduct sections. Flattenedepitheliumand lumen widening of collecting ducts inhomozygous kidneys (M) compared with wild-type animals (L). Note the expansion of thestroma component between ducts (*) (PAS).(M–O) EM analysis of mature glomeruli. Po-docyte foot process effacement (arrowheads)and segmental duplication of GBM (arrows) inhomozygous mutant newborn P0 kidneys (N)contrasting with normal architecture of po-docytes and GBM in wild-type littermates.Note the presence of large intercellular junc-tions between podocytes in homozygousmutant kidneys (white arrow) (P). Scalebars: (DandE):500mm; (F–I,K): 50mm; (LandM):20 mm; (J) 100 mm; (N and O): 1 mm; and (O):25 nm.
J Am Soc Nephrol 27: 1042–1054, 2016 Renal Defects in HANAC Col4a1 Mice 1043
www.jasn.org BASIC RESEARCH
renal disease that associates multicystic kidneys, sometimes he-maturia, and decreased GFR in older patients.17–19 HANACCOL4A1 mutations all substitute glycine residues between
Gly498 and Gly528 within the cyanogenbromide–derived fragment CB3[IV] ofthe protein that contains integrin-bindingsites.16–18 These data suggest that thepathogenic effects of HANAC mutationsmay occur through defective cell–BM in-teractions. Because COL4A1mutations as-sociated with the brain and eye phenotypeare usually localized in the C-terminal halfof the protein, phenotype-genotype correla-tions may occur among COL4A1-relateddiseases.
Because kidney disease, including cystsand hematuria, has not been reported inpatients with brain-eye restricted dis-ease and in ENU Col4a1 mice,7,14,15 wegenerated mice harboring the Col4a1p.Gly498Val mutation previously identifiedin a family with HANAC to specifically in-vestigate the pathophysiology of renaldefects.
RESULTS
Generation of HANAC Col4a1 G498VMutant MiceA targeting vector was designed to intro-duce the Col4a1 c.G1617T mutation inexon 25 leading to the p.Gly498Val substi-tution (G498V) (Supplemental Figure 1A).Electroporation of the construct intomouse embryonic stem (ES) cells resultedin 372 neomycin-resistant clones of whichfive were targeted (Supplemental Figure1B). Three positive clones were injectedinto mouse blastocysts, and four chimericmice were obtained. The targeted allele wastransmitted through the germline (Supple-mental Figure 1, C and D). Crossbreedingof Col4a1+/G498V mice generated 32% ofCol4a1+/+, 51% of Col4a1+/G498V, and17% of Col4a1G498V/G498V, indicatingincreased embryonic lethality of the ho-mozygous mutants (expected Mendeliandistribution 25%, 50%, and 25%, respec-tively). After birth however, heterozygousand homozygous mice showed normal vi-ability. Brain hemorrhages were observedin Col4a1+/G498V and Col4a1G498V/G498V
newborns that did not impair life span(data not shown), and retinal tortuosity
and muscular dystrophy were present in adult animals(A. Trouillet et al., submitted; S. Guiraud et al., manuscriptin preparation).
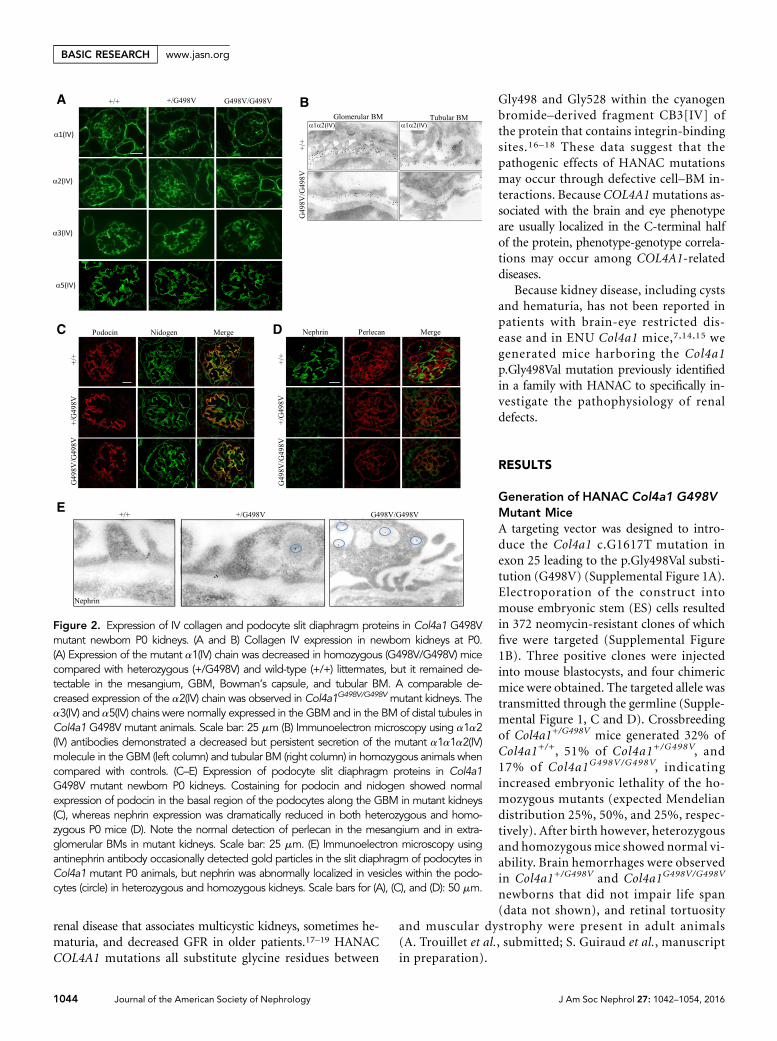
Figure 2. Expression of IV collagen and podocyte slit diaphragm proteins in Col4a1 G498Vmutant newborn P0 kidneys. (A and B) Collagen IV expression in newborn kidneys at P0.(A) Expression of the mutant a1(IV) chain was decreased in homozygous (G498V/G498V) micecompared with heterozygous (+/G498V) and wild-type (+/+) littermates, but it remained de-tectable in the mesangium, GBM, Bowman’s capsule, and tubular BM. A comparable de-creased expression of the a2(IV) chain was observed in Col4a1G498V/G498V mutant kidneys. Thea3(IV) and a5(IV) chains were normally expressed in the GBM and in the BM of distal tubules inCol4a1 G498V mutant animals. Scale bar: 25 mm (B) Immunoelectron microscopy using a1a2(IV) antibodies demonstrated a decreased but persistent secretion of the mutant a1a1a2(IV)molecule in the GBM (left column) and tubular BM (right column) in homozygous animals whencompared with controls. (C–E) Expression of podocyte slit diaphragm proteins in Col4a1G498V mutant newborn P0 kidneys. Costaining for podocin and nidogen showed normalexpression of podocin in the basal region of the podocytes along the GBM in mutant kidneys(C), whereas nephrin expression was dramatically reduced in both heterozygous and homo-zygous P0 mice (D). Note the normal detection of perlecan in the mesangium and in extra-glomerular BMs in mutant kidneys. Scale bar: 25 mm. (E) Immunoelectron microscopy usingantinephrin antibody occasionally detected gold particles in the slit diaphragm of podocytes inCol4a1 mutant P0 animals, but nephrin was abnormally localized in vesicles within the podo-cytes (circle) in heterozygous and homozygous kidneys. Scale bars for (A), (C), and (D): 50 mm.
1044 Journal of the American Society of Nephrology J Am Soc Nephrol 27: 1042–1054, 2016
BASIC RESEARCH www.jasn.org

Col4a1 G498V Mutant Newborn MiceShowed Delayed GlomerulogenesisResulting in GlomerularHyperpermeability at BirthWe previously reported that the COL4A1G498V mutation is responsible for hematu-ria and multicystic kidney disease.16,18
Col4a1+/G498VandCol4a1G498V/G498V newbornsat postnatal day 0 (P0) showed hematuria,with numerous urinary dysmorphic eryth-rocytes suggesting their glomerular origin(Figure 1, A and B). Albuminuria was alsodetected in mutant newborns, with meanurinary albumin concentrations of 34.667.8, 141.16129.4, and 942.06507.5 mg/l(P,0.001) in wild-type, heterozygous, andhomozygous P0 mice, respectively (Figure1C). Compared with the normal morphol-ogy of wild-type animals, homozygouskidneys showed alterations of glomeruliand tubules at P0 (Figure 1, D and E). De-fective glomerulogenesis was observed af-ter the formation of the first capillaryloops, with podocytes arranging in multi-layers instead of forming a regular layer(Figure 1, F and G). Additionally, in a sub-set of mature glomeruli, Bowman’s spaceappeared enlarged with retracted capillarytufts and widened capillary loops, withoutmesangiolysis (Figure 1, H–J). Proximaltubular sections in Col4a1+/G498V (notshown) and Col4a1G498V/G498V (Figure1K) newborn kidneys showed cytoplasmicvacuoles that positively stained with anti-albumin antibody (data not shown), mostlikely linked to proteinuria. Col4a1G498V/G498V
kidneys also showed focal dilation withflattened proximal tubule cells (Figure 1J).Collecting duct sections showedflattened ep-ithelium with lumen widening, and ductswere focally dissociated by expansion ofstroma (Figure 1, E and M).
Electronmicroscopy (EM)of deep corticalglomeruli in homozygous kidneys at P0revealed extensive foot process effacementwith focal persistence of large cell cell-junctionbetween podocytes and areas of splitting andduplication of the GBM (Figure 1, N–P).Although the glomerular architecture ap-peared intact in heterozygous kidneys, re-duced podocyte foot process formation andGBM duplication were also observed by EM(data not shown).
During the first week, albuminuria de-creased, with recovery of the morphologic
Figure 3. Morphologic and functional characteristics of Col4a1 G498V mutant adultkidney. (A) Histologic analysis of 6-month adult kidney. Heterozygous (+/G498V) and ho-mozygous (G498V/G498V) mice developed cystic glomeruli with dilation of Bowman’sspace and retracted capillary tuft (first column, Masson’s trichrome). Scale bars: 200 mm.Noncystic glomeruli showed cuboidal PECs (black arrows) (second column, Jones’ me-thenamine silver). Nodular inflammatory infiltrates were seen in both heterozygous andhomozygous mutant kidneys around glomeruli (third column) and arterioles (fourth column)(Masson’s trichrome). Scale bar: 50 mm. (B) Transverse sections of 12-month heterozygousand homozygous kidneys showing distribution of glomerular cysts throughout the cortexarea. Note the dysmorphic papilla in homozygous kidney (Masson’s trichrome). Scale bar:1 mm. (C) Glomeruli with cystic change (cyst surface, left panel: 11,283 mm2; cyst surface,right panel: 14,900 mm2) showing a normal glomerulotubular junction (black arrow) in+/G498V and G498V/G498V 6-month kidneys (periodic acid–Schiff). Scale bar: 20 mm.(D) Percentage of glomeruli with cystic changes (left panel) and glomerular cyst area (rightpanel) in heterozygous and homozygous kidneys at 3 months (+/G498V, n=4; G498V/G498V, n=4), 6 months (+/G498V, n=8; G498V/G498V, n=6) and 12 months (+/G498V,n=7; G498V/G498V, n=7) of age. Values represent mean6SEM. *P,0.05, ***P,0.001.
J Am Soc Nephrol 27: 1042–1054, 2016 Renal Defects in HANAC Col4a1 Mice 1045
www.jasn.org BASIC RESEARCH

glomerular and tubular lesions (Supplemental Figure 2, A andB).Erythrocyte casts remained present in tubular sections of 7-day-oldmutant kidneys (Supplemental Figure 2C), but they were notobserved at 1month. In addition, neonate kidney defects did notresult in a decreased glomerular density in adult Col4a1 G498Vmutant animals (Supplemental Figure 2D).
Altogether, these results indicate that the Col4a1 G498Vmutation induced delayed glomerulogenesis with defectiveGBM and podocyte maturation, resulting in glomerular hy-perpermeability in newborn animals.
Col4a1 G498V Mutation Decreased a1a1a2(IV) TrimerSecretion and Altered Nephrin Expression in NewbornP0 KidneysTo assess the effect of the Col4a1 G498Vmutation on type IV col-lagen expression, we analyzed the renal distribution of thea1a1a2(IV) trimer. In Col4a1G498V/G498V newborn kidneys, the expressionof the mutant a1a1a2(IV) trimer was reduced compared withwild-type and heterozygous mice, but it remained detectable inthemesangium, Bowman’s capsule, tubular BM, andGBM (Figure2, A andB), which indicated that themutation only partially altereda1a1a2(IV) secretion. GBMexpression of the a3(IV) and a5(IV)chains were comparable between genotypes (Figure 2A).
Given the podocyte defects observed in the mutant newbornsat P0, we investigated the expression of slit diaphragm proteins.Although the expression of podocin remained normal (Figure2C), that of nephrin was dramatically reduced in Col4a1+/G498V
and Col4a1G498V/G498V glomeruli (Figure 2D). By immunogoldEM, nephrinwas occasionally detected in slit diaphragmswhile itwas accumulated in cytoplasmic vacuoles (Figure 2E). Defectivenephrin expressionwas consistentwith the dramatic reduction offoot process formation and albuminuria in P0 Col4a1 mutants.
Col4a1 G498V Mutant Adult Mice DevelopedGlomerulocystic Kidney Disease and DysmorphicPapillae with Mild Renal Function AlterationAlthough tubular sections were normal in 1-month mutantkidneys, careful analysis disclosed abnormal morphology of theglomerularparietal epithelial cells (PECs) inapproximately15%ofmutant glomeruli. PECs were cube-shaped with enlarged nuclei,instead of forming a monolayer of thin epithelial cells with flatnuclei (Supplemental Figure 3A). At 3 months of age, glomerularcysts and inflammatory infiltratesfirst developed inCol4a1+/G498V
andCol4a1G498V/G498V kidneys. Glomerular cysts were distributedthroughout the cortex area and were characterized by enlargedBowman’s space lined by flattened PECs and retracted capillarytufts (Figure 3, A and B). Glomerulotubular junction was pre-served in normal glomeruli and in glomeruli undergoing cystictransformation (Figure 3C). The percentage of glomeruli withcystic changes significantly increased at 6 months in both hetero-zygous and homozygous mice, and also at 12 months in homo-zygous mutants (Figure 3D). Cystic surfaces were comparablebetween ages and genotypes, except at 12 months where glomer-ular cysts appeared slightly but significantly larger in heterozygouskidneys (Figure 3D). Cuboidal PECs lining the Bowman’s capsule
were present in about one-third of noncystic glomeruli in 6- and12-month animals (Figure 3A). Large infiltratesmostly composedof CD3-positive lymphocytes surrounded by F4/80-positivemac-rophages occasionally developed around cystic and noncystic glo-meruli and around arterioles or venules with intact vessel walls(Figure 3A, Supplemental Figure 3, B and C). No significant fi-brosis developed except in the inflammatory areas, and tubulesremained normal at 12 months.
Ultrastructural analysis of adult mutant kidneys performedat 6 months disclosed morphologic PEC alterations, withthickening and multilamination of the Bowman’s capsule(Supplemental Figure 3D), whereas the GBM and the tubularBM remained normal (not shown).
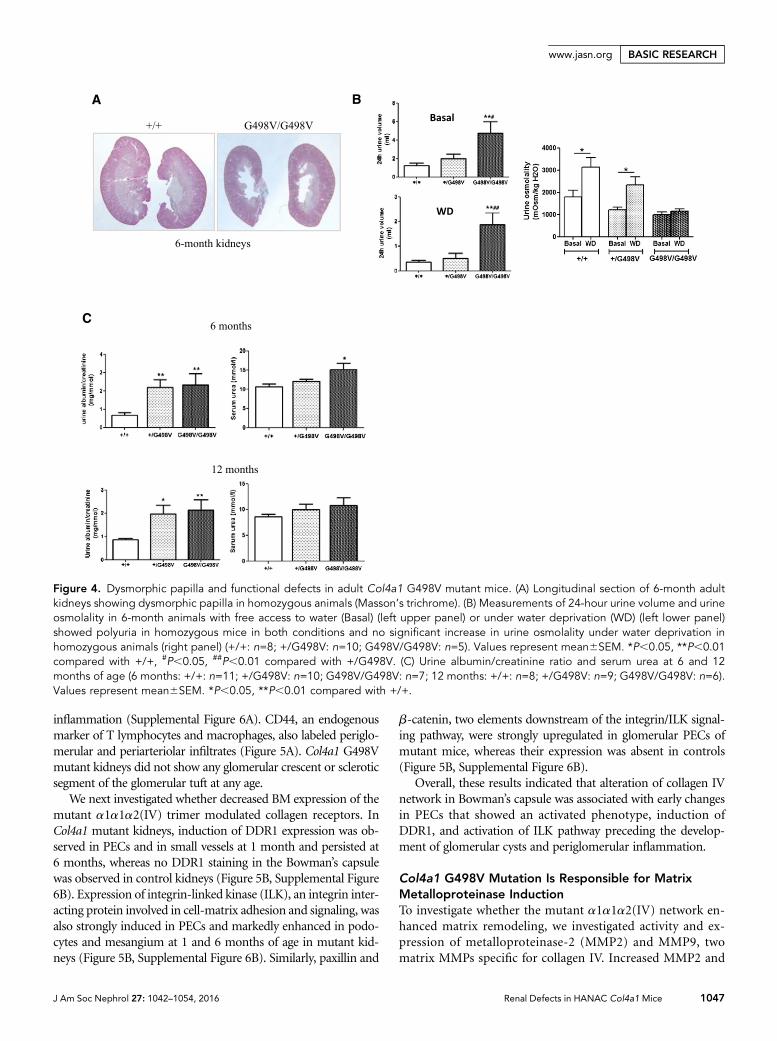
Beside glomerular and vascular alterations, homozygousmice showed dysmorphic papillae (Figure 4A), which wereassociated with polyuria and defects in urine concentrationunder water deprivation (Figure 4B).
As previously observed in newborn kidneys,a1(IV) and a2(IV) chain staining analyzed at 6 months was decreased in themesangium and tubular BM of homozygous adults and in theBowman’s capsule where the expression of the a5a5a6(IV)trimers remained normal (Supplemental Figure 4A). Interme-diate staining was observed in heterozygous mutants. Expres-sion of the BM proteins, nidogen/entactin, laminin a2, andlaminin b1, was normal, as was that of fibronectin and colla-gen I (Supplemental Figure 4B).
Renal functional parameters were assessed at 6 and 12monthsof age. Urine albumin/creatinine was mildly but significantlyincreased in6-and12-month-oldmutantanimals comparedwithcontrol littermates (Figure 4C). Only homozygous mutantsshowed an increase of serum urea at 6 months (10.760.66,12.160.51, and 15.160.16 mmol/l in Col4a1+/+, Col4a1+/G498V,and Col4a1G498V/G498V, respectively, P,0.05; Col4a1G498V/G498V
compared with controls), whereas at 12 months, urea was non-significantly increased in this genotype (Figure 4C). Becausebody weights at 6 and 12 months were significantly lower inCol4a1G498V/G498Vanimals (Supplemental Figure 5), these resultssuggest that alteration of kidney function was likely underesti-mated in homozygous mutant animals.
Col4a1 G498V Mutation Led to PEC Activation andInduction of DDR1 and Integrin-Linked Kinase PathwayWefocusedon thealterationsof thePECs in6-monthanimalswithovert cystic renal disease. The tight junction protein claudin-1, aspecific marker of PECs, was strongly expressed at the apical andlateral surface of PECs inmutantmice,whereas itwas observed at alower level in control kidneys (Figure 5A). More importantly,cuboidal PECs expressed CD44, a marker of activated parietalepithelial cells (aPECs), not detected in normal glomeruli or innormal flat-shape PECs in mutant kidneys (Figure 5A). PECs lin-ing cystic glomeruli showed de novo expression ofa-smoothmus-cle actin (a-SMA), whereas cube-shaped PECs were rarely stainedfora-SMA (Figure 5A). Claudin-1 upregulation andCD44 induc-tion in PECs were actually first detected in 1-month-old micebefore thedevelopmentof the glomerular cysts andperiglomerular
1046 Journal of the American Society of Nephrology J Am Soc Nephrol 27: 1042–1054, 2016
BASIC RESEARCH www.jasn.org
inflammation (Supplemental Figure 6A). CD44, an endogenousmarker of T lymphocytes and macrophages, also labeled periglo-merular and periarteriolar infiltrates (Figure 5A). Col4a1 G498Vmutant kidneys did not show any glomerular crescent or scleroticsegment of the glomerular tuft at any age.
We next investigated whether decreased BM expression of themutant a1a1a2(IV) trimer modulated collagen receptors. InCol4a1 mutant kidneys, induction of DDR1 expression was ob-served in PECs and in small vessels at 1 month and persisted at6 months, whereas no DDR1 staining in the Bowman’s capsulewas observed in control kidneys (Figure 5B, Supplemental Figure6B). Expression of integrin-linked kinase (ILK), an integrin inter-acting protein involved in cell-matrix adhesion and signaling, wasalso strongly induced in PECs and markedly enhanced in podo-cytes and mesangium at 1 and 6 months of age in mutant kid-neys (Figure 5B, Supplemental Figure 6B). Similarly, paxillin and
b-catenin, two elements downstream of the integrin/ILK signal-ing pathway, were strongly upregulated in glomerular PECs ofmutant mice, whereas their expression was absent in controls(Figure 5B, Supplemental Figure 6B).
Overall, these results indicated that alteration of collagen IVnetwork in Bowman’s capsule was associated with early changesin PECs that showed an activated phenotype, induction ofDDR1, and activation of ILK pathway preceding the develop-ment of glomerular cysts and periglomerular inflammation.
Col4a1 G498V Mutation Is Responsible for MatrixMetalloproteinase InductionTo investigate whether the mutant a1a1a2(IV) network en-hanced matrix remodeling, we investigated activity and ex-pression of metalloproteinase-2 (MMP2) and MMP9, twomatrix MMPs specific for collagen IV. Increased MMP2 and
Figure 4. Dysmorphic papilla and functional defects in adult Col4a1 G498V mutant mice. (A) Longitudinal section of 6-month adultkidneys showing dysmorphic papilla in homozygous animals (Masson’s trichrome). (B) Measurements of 24-hour urine volume and urineosmolality in 6-month animals with free access to water (Basal) (left upper panel) or under water deprivation (WD) (left lower panel)showed polyuria in homozygous mice in both conditions and no significant increase in urine osmolality under water deprivation inhomozygous animals (right panel) (+/+: n=8; +/G498V: n=10; G498V/G498V: n=5). Values represent mean6SEM. *P,0.05, **P,0.01compared with +/+, #P,0.05, ##P,0.01 compared with +/G498V. (C) Urine albumin/creatinine ratio and serum urea at 6 and 12months of age (6 months: +/+: n=11; +/G498V: n=10; G498V/G498V: n=7; 12 months: +/+: n=8; +/G498V: n=9; G498V/G498V: n=6).Values represent mean6SEM. *P,0.05, **P,0.01 compared with +/+.
J Am Soc Nephrol 27: 1042–1054, 2016 Renal Defects in HANAC Col4a1 Mice 1047
www.jasn.org BASIC RESEARCH

MMP9 activity was detected in both heterozygous and homo-zygous kidney homogenates by SDS-PAGE gelatin zymogra-phy at 6 months (Figure 6A). RT-PCR experiments showedthat mRNA of MMP2 and MT1-MMP involved in MMP2activation was upregulated in 6-month Col4a1 mutant
kidneys, whereas MMP9 mRNA was only significantly in-creased in heterozygous mice (Figure 6B). Likewise, mRNAlevels of tissue inhibitors of MMPs TIMP1 and TIMP2 wereincreased (Figure 5B). We then performed in situ zymography,which localized increased gelatinase activity to the Bowman’s
Figure 5. Phenotypic alterations of glomerular PECs in Col4a1 G498V mutant adult mice. (A) Immunohistochemical staining ofa 6-month adult kidney with antibodies to claudin-1, CD44, and a-SMA (left panels). In heterozygous (+/G498V) and homozygous (G498V/G498V) mutants, cuboid shape PECs in noncystic glomeruli showed claudin-1 upregulation and induction of CD44 expression. PECs liningcystic glomeruli stained positive for a-SMA. Right panels illustrate the selective induction of CD44 marker in cuboid-shape PECs (upper rightpanel) and absence of CD44 expression in normal flat-shape PECs (lower right panel) in 6-month mutant glomeruli. Note the presence ofperiglomerular inflammatory cells that also are stained for CD44. Scale bar: 50 mm. (B) Expression of the DDR1, ILK, paxillin, and b-catenin ina 6-month adult kidney. DDR1 expression was induced in the Bowman’s capsule and the small vessel wall of Col4a1+/G498V and Col4a1G498V/G498V
mice. ILK and ILK downstream partners, paxillin, and b-catenin were induced in PECs. In addition, ILK expression was strongly enhancedin the mesangium and podocytes in both mutant genotypes compared with controls (+/+). Scale bars: 50 mm.
1048 Journal of the American Society of Nephrology J Am Soc Nephrol 27: 1042–1054, 2016
BASIC RESEARCH www.jasn.org

capsule and the tubular BM in 6-month mutant kidneys. Theenzymatic activity was inhibited by EDTA, indicating thatMMP2 and/or MMP9 carried gelatinolytic activity (Figure6C). These results showed that despite increased MMP inhib-itor mRNAs, the resulting gelatinolytic activity was increasedin mutant kidney tissue, particularly in the Bowman’s capsule.
DISCUSSION
Generation of the first animal model of HANAC syndromeprovided us the opportunity to analyze the pathophysiology oforgan defects in this multisystemic syndrome and to approachthe pleiotropic role of the ubiquitously expresseda1 isoformofcollagen IV.
SeveralCol4a1ENUmutantmice have been analyzed.7,9,13–15,20
They usually showed severe brain disease and eye abnormalities,but no consistent kidney alteration has been reported so far, espe-cially renal cysts, a cardinal phenotypic characteristic ofHANAC.16,18 Because HANAC COL4A1 mutations are localizedwithin a 30 amino acid segment containing integrin-binding sites,alteration of this protein domainmight trigger specific pathogenicpathways.
Contrasting with previous ENU Col4a1 animals, Col4a1G498V homozygous newborns are viable. The detection ofthe mutant a1a1a2(IV) trimer in BMs of homozygous ani-mals may account for the less severe phenotype observed inour Col4a1 mutant animals. Indeed, in homozygous ENUCol4a1mice with exon 40 deletion, thea1(IV) chainmutationresults in the absence of the collagen IV network in embryonicBMs responsible for embryonic lethality.7,13 Therefore, as pre-viously demonstrated in Alport syndrome,21–23 the type ofmutation and location of glycine substitution may influencethe severity of the phenotype in COL4A1-related diseases.
Col4a1G498Vmutantmice are a relevant animal model forHANAC, reproducing the organs defects observed in patients,including retinal tortuosity and muscular dystrophy(A. Trouillet et al., manuscript submitted; S. Guiraud et al.,manuscript in preparation), brain hemorrhages, and renalcysts in adults. However, we also unexpectedly observed renalalterations in Col4a1G498Vmutant newborns. Increased glo-merular permeability observed in these animals is related todelayed podocyte differentiation, nephrin mislocalization,and GBM disorganization that resolve during the first weeksafter birth. The a1a1a2(IV) trimer constitutes the first colla-gen IV network of the embryonic GBM,24 and we provide thedemonstration that it contributes to podocyte maturation. In-terestingly, podocyte and GBM alterations are reminiscent ofthe findings in mice lacking the a3 integrin and in those withpodocyte-specific deletion of the b1 integrin.25,26 Immaturecollecting ducts in Col4a1 G498V mutant mice at birth werealso observed in a3 integrin defective mice.25 During nephro-genesis, the a3b1 integrin is expressed in the ureteric budand in podocyte precursors.27,28 It specifically localizes inthe podocyte foot processes and contributes to podocyte
cytoskeleton stabilization through interaction with GBMcomponents. The GBM ligand for the a3b1 integrin in matureglomerulus is laminin 521,29 but the ligand is unknown inearlier stage of glomerulogenesis. Data in our mutant micewith delayed podocyte maturation suggest that the ligandmight be a1a1a2(IV). This hypothesis is supported by thesite of the Col4a1 G498V substitution, which alters an integrin-binding motif.18 Additionally, reduced integration of thenormal a1(IV) chain and/or expression of the mutant a1(IV)isoform in the collagen IV network might weaken the GBM.The replacement of embryonic isoforms of collagen IV andlaminin, by a3a4a5(IV) and laminin 521, respectively,30 mayaccount for morphologic and functional recovery of the glo-merular filtration barrier in 1-month animals. Finally, the de-layed glomerulogenesis observed in the mutant newborns didnot affect the nephronic mass because glomerular density andkidney weights were preserved in adult kidneys.
Analysis of the adult Col4a1 G498V mutant kidneys pro-vided new insights into the cystic kidney phenotype observedin patients with HANAC. Actually, computed tomographyscan imaging did not allow identification of the origin of therenal cysts, which showed a broad cortical distribution in pa-tients with HANAC syndrome,16 a pattern that remains com-patible with a glomerulocystic kidney disease.31 Kidney biopsyspecimens were analyzed in two young patients with normalrenal imaging and did not identify cyst formations.16,17 His-tologic observations in adult mutant animals now clearly showthat cysts arise from glomeruli with Bowman’s space dilatationand they distribute through the cortex area. As observed in sev-eral patients, the number of cysts increased with age in mutantmice. Therefore, HANAC potentially represents a new inheritedformof glomerulocystic kidney disease, which adds to autosomaldominant or recessive polycystic kidney disease and uromodulinand HNF1b/TCF2 mutation–related disorders.31
Complex alterations of the Bowman’s capsule and the PECsprecede glomerular cysts in mutant adult kidneys. PECs fea-ture morphologic characteristics of activated PECs with coex-pression of claudin-1 and CD44.32–34 These alterations wereseen as early as 1 month before appearance of the glomerularcysts and periglomerular inflammation. PEC phenotypicalchanges were not associated either with glomerular crescentor pseudocrescent formations, as observed in crescentic glo-merulonephritis and collapsing glomerulopathy, respectively,or lesions of segmental glomerulosclerosis.32–34
In glomerular diseases associatedwith the aPECphenotype,triggers of PEC activation remain poorly defined. Interestingly,aPECs in Col4a1 G498V mutant mice show induction ofDDR1 and the ILK signaling pathway. DDR1 links collagenIV and plays an important role in cell response to matrix re-modeling and proinflammatory response.35–38 In our mice,DDR1 induction may be the consequence of the enhancedmatrix turnover observed in the Bowman’s capsule, as shownby increased collagenase activity and ultrastructural altera-tions. Periglomerular inflammation may result from DDR1overexpression, which may also account for perivascular
J Am Soc Nephrol 27: 1042–1054, 2016 Renal Defects in HANAC Col4a1 Mice 1049
www.jasn.org BASIC RESEARCH

Figure 6. Matrix MMP induction in Col4a1 G498V mutant adult kidneys. (A) SDS-PAGE zymography of protein extracts from 6-monthkidneys showed induction of pro-MMP9 and upregulation of pro-MMP2 and MMP2 enzymatic activities in Col4a1+/G498V andCol4a1G498V/G498V mutant kidneys compared with control littermates (+/+). (B) Renal mRNA level of MMP-2, MMP-9, MT1-MMP,TIMP1, and TIMP2 (+/+: n=5; +/G498V: n=5; G498V/G498V: n=3) in 6-month adult kidneys. Values represent mean6SEM. *P,0.05compared with +/+. (C) In situ gelatin zymography revealed enhanced MMP enzymatic activity in the periglomerular region and aroundthe renal tubules in 6-month-old Col4a1+/G498V and Col4a1G498V/G498V kidneys. Pretreatment of the renal tissue with EDTA, a specificinhibitor of MMP2 and MMP9, blocked gelanolytic activity. Scale bar: 100 mm.
1050 Journal of the American Society of Nephrology J Am Soc Nephrol 27: 1042–1054, 2016
BASIC RESEARCH www.jasn.org

inflammation because upregulation of DDR1 was observed inthe vascular wall.
Although activation of ILK pathway is well established inpodocytes,39–41 it was not previously reported in PECs. ILKregulates extracellular matrix-induced signaling pathwaythrough binding of b1 and b3 integrins.42 In our mousemodel, induction of ILK in PECs may result from an en-hanced, integrin-mediated signaling pathway triggered byCol4a1 mutation and/or collagen IV network alteration. Inturn, ILK activationmay account forb-catenin overexpressionthrough inhibition of GSK3b, which promotes b-catenin deg-radation in normal conditions.43We have shown upregulationof claudin-1 and induction of CD44, which are both targetgenes for b-catenin/Tcf(Lef-1) pathways.44,45
Mechanisms of glomerular cyst formation have not beenpreviously addressed. We have not observed morphologicdefects that would support the hypothesis of obstructivenephropathy as explanation for the glomerulocystic features.In particular, glomerulotubular junction was preserved inglomeruli with cystic change. The mutated Col4a1 protein,together with decreased a1a1a2(IV) trimer secretion and en-hanced matrix remodeling, may alter the mechanical proper-ties of the Bowman’s capsule and may therefore inefficientlycounteract the glomerular filtration pressure. In addition,there are dramatic alterations of PECs that show signs of ac-tivation at 1 month and later on show signs of mesenchymaltransition in PECs lining glomerular cysts, with expression ofa-SMA. These alterations might be related to activation ofILK, which has been shown to promote epithelial mesen-chymal transition46–48 and enhance MMP2 and MMP9activity.49,50
In addition to the complex glomerular alterations observedinCol4a1G498Vmutant mice, dysmorphic papillae and urineconcentration defects were selectively observed in adulthomozygous animals, as were collecting duct anomalies inhomozygous neonates. Such functional alterations were notrecorded in patients with HANAC, which carry however theCOL4A1 mutation in the heterozygous state.
In summary, our study reveals the previously unrecognizedrole during glomerulogenesis ofa1a1a2(IV), thefirst collagenIV network of the GBM, and its contribution to podocytedifferentiation. In mature kidneys, a1a1a2(IV) collagen net-work integrity is crucial for maintenance of PEC differentia-tion. Primary molecular alterations of the Bowman’s capsulematrix trigger PEC activation and mesenchymal transition.From a clinical point of view the Col4a1 G498V mutantmice provide new insight into the pathomechanisms of glo-merular cystic kidney disease.
CONCISE METHODS
Generation of Col4a1 G498V Mutant MiceThe Col4a1 G498V mutant mouse line was established at the MCI/ICS
(Mouse Clinical Institute, Illkirch, France; http://www-mci.u-strasbg.fr).
A 4.8-kb fragment encompassing introns 22–26 and exons 22–25
of the Col4a1 gene was amplified by PCR usingmus musculus DNA
as a template and subcloned in an MCI proprietary vector, result-
ing in step1 plasmid. This MCI vector has a floxed neomycin re-
sistance cassette driven by a phosphoglycerate kinase 1 promoter.
A 3.1-kb fragment encompassing intron 26 was amplified by
PCR and subcloned in step1 plasmid to generate the final targeting
construct. The Col4a1 c.G1617Tmutation was introduced in exon 25
via site-directed mutagenesis. Sequencing of the final targeting vector
was performed. The linearized targeting construct was electroporated
in 129Sv/Pas mouse ES cells. After selection, five targeted clones were
identified by PCR using external primers and confirmed by Southern
blot using 59 and 39 external probes (Supplemental Figure 1). Two
positive ES clones were injected into C57BL/6N blastocysts, and the
derived male chimeras gave germline transmission. Tail biopsies from
F1 mice were genotyped with primers flanking neomycin cassette.
Col4a1+/G498V offsprings were crossed with CMV-Cre mice. Off-
springs were assessed by PCR for the excision of the cassette, and
several heterozygous Col4a1+/G4984V were obtained. Col4a1+/G498V
mice on mixed background were crossed with wild-type mice from
C57BL/6 background for two generations. Genotyping was per-
formed by PCR using Ef/Er primers flanking the residual Lox site,
and presence of the p.Gly498Val was confirmed by sequencing of
genomic DNA from Col4a1+/G498V animals (Supplemental Figure
1). Sex of newborns was determined by PCR amplification of geno-
mic DNA sequence of the Kdm5c gene.51 Newborn mice were eutha-
nized. Adult mice were euthanized at 7 days, 1 month, 3 months, 6
months, and 1 year with intraperitoneal administration of pentobar-
bital (150 mg/kg). Mice were maintained in a specific pathogen-free
environment, and experiments were conducted in accordance with
French government policies.
Histologic and Ultrastructural AnalysisFor light microscopy analysis, kidneys were fixed with alcohol-
formalin-acetic acid (AFA) solution, dehydrated, and embedded in
paraffin. There were 4-mm sections that were deparaffinized, rehy-
drated, and stained with Masson’s trichrome, hematoxylin and eosin,
and periodic acid–Schiff. For quantification of glomerular density,
glomerular cyst number, and size, longitudinal kidney sections were
scanned at a high resolution using a slide scanner (NanoZoomer 2.0-
HT; Hamamatsu, Massy, France). Size of glomerular cysts was mea-
sured on at least two different section levels/kidneys using Image J
software. Glomeruli were counted one by one, using the Nano-
Zoomer software, within cortical areas of 1-mm depth from the renal
capsule that covered the kidney sections; glomerular density was eval-
uated as the average number of glomeruli per square millimeter.
Transmission EM was performed on kidney samples fixed in 2.5%
glutaraldehyde dissolved in 0.1 mol/L cacodylate buffer (pH 7.4) at
4°C. The tissue fragments were postfixed in 1%OsO4, dehydrated, and
embedded in epoxy resin. Semi-thin sections (0.5mm)were stained by
toluidine blue, and 60-nm ultra-thin sections were contrasted with
electron-opaque solution uranyl acetate and then lead citrate; they
were then examined using a JEOL 1010 electron microscope (JEOL,
Tokyo, Japan) and a MegaView III camera (Olympus Soft Imaging
Systems, Münster, Germany).
J Am Soc Nephrol 27: 1042–1054, 2016 Renal Defects in HANAC Col4a1 Mice 1051
www.jasn.org BASIC RESEARCH

Immunohistochemistry, Immunofluorescence, andImmunoelectron MicroscopyAFA-fixed, paraffin-embedded sections were used for immunohis-
tochemistry. Antigen retrieval was performed by heat in citrate buffer
(pH 6.0) or with proteinase K/PBS solution. Tissue sections were
probed using primary antibodies, followed by incubation with
Histofine immunohistochemical staining reagent (Nichirei Biosci-
ences, Japan). Immunofluorescence experiments were carried out
with 4-mm cryosections or AFA-fixed, paraffin-embedded sections of
kidney, depending on the primary antibody affinity. Following block-
ade by 10% BSA/PBS solution, the sections were incubated with
primary antibodies overnight. The slides were then incubated with
appropriate AlexaFluor 488– or AlexaFluor 555–labeled secondary
antibodies (Invitrogen) for 1 hour at room temperature. Immuno-
fluorescence micrographs were obtained using a Leica TCS SP2 con-
focal microscope. Primary antibodies used for immunostaining are
listed in Supplemental Table 1. Immunogold EM was performed on
ultrathin frozen sections of kidney tissue as previously described.52
Sections were processed for indirect immunogold labeling with the
use of rabbit anti–a1a2(IV) collagen (Rockland) and guinea pig anti-
nephrin (PROGEN).
Urine and Blood Samples AnalysisUrine samples were collected usingmetabolic cages in adultmicewith
free access to water or after 24 hours of water deprivation. Peripheral
blood was collected after puncture of the retro-orbital sinus in mice
anesthetized using pentobarbital 10%. Urine osmolality was mea-
sured using aVapro 5520 vapor pressure osmometer (Wescor). Serum
urea, urine creatinine, and albuminuria were measured by photom-
etry using an Olympus AU 400 apparatus (IFR02; Hôpital Bichat,
Paris, France). Urine of newborn mice (P0–P7) was collected by
spot urine sampling. For SDS-PAGE analysis, 1 ml of urine sample
was loaded on polyacrylamide gel electrophoresis, and the gels then
were stained with Coomassie Brilliant Blue solution.
Detection of Matrix MMP ActivityGelatinolytic activity was determined by in situ zymography on 8-mm
unfixed cryostat sections using quenched fluorogenic substrate DQ
gelatin (Molecular Probes). Then 1mg/ml of DQ gelatin solutionwas
prepared with water and was 1:10 diluted in 1% (w/v) low gelling
temperature agarose (Sigma-Aldrich) in PBS. The 50-ml samples of
mixture were applied on top of air-dried cryosections and covered
with a coverslip and gelled after incubation at 4°C.53 The slides were
incubated for 3 hours at 37°C, and fluorescence was analyzed under
confocal microscopy. To determine the specificity of gelatinolytic
activity, kidney cryosections were preincubated for 1 hour at room
temperature with 20-mM EDTA in PBS, which inhibits Mmp2 and
Mmp9 activity, and incubation with DQ gelatin was performed as
previously described.
SDS-PAGE gelatin zymography was performed with renal tissue
homogenized in RIPA lysis buffer supplemented with PMSF, sodium
orthovanadate, and protease inhibitor cocktail solution (sc-24948;
Santa Cruz Biotechnology) using a T10 Basic Ultra-Turrax disperser
(IKA, Germany). Gelatin zymography was performed with tissue
homogenates using 8% polyacrylamide gel containing 1 mg/ml of
gelatin. After electrophoresis, gels were washed in 2.5% Triton X-100
and then incubated in ZnCl2/CaCl2 containing Tris buffer. As a
negative control, identical gels were incubated in EDTA-containing
Tris buffer. Gels were then stained in Coomassie Brilliant Blue
solution.
Quantitative Real-Time PCRTotal RNAs were prepared from kidney tissue samples using the
TRIzol Reagent (Life Technologies) and the T10 Basic Ultra-Turrax
disperser. RNA quality was checked by NanoDrop, and residual
genomic DNA was removed by DNase I treatment (Fermentas). For
quantitative reverse-transcription polymerase chain reaction, the
mRNAwas converted into cDNA using the Revert Aid H minus First
Strand DNA Synthesis kit (Fermentas). cDNAwas amplified by PCR
using specific primers, FastStartDNAMaster SYBRGreen I kit (Roche
Diagnostic) and the LightCycler 480 System (Roche Diagnostic). For
the normalization of gene expression data, the GeNorm was used to
determine the two most stable references among five candidate
housekeeping genes. Results are presented as normalized relative ex-
pression levels.
Statistical AnalysesAll data are presented as individual measurements with mean6SEM.
Statistical significance was determined by using either nonparametric
tests, including Mann–Whitney and Kruskal–Wallis tests, or the
paired t test, when appropriate. All statistical tests were calculated
using GrapPad Prism 5.00 (GraphPad Software, San Diago, CA). Sta-
tistical significance was defined by P,0.05, P,0.01, and P,0.001.
Study ApprovalAll of the experimental procedures were approved by the ethics
committee at the University Pierre et Marie Curie and the French
Ministry of Research (agreement 01108.01) and were performed in
accordance with the National Institutes of Health Guide for the Care
and Use of Laboratory Animals and the French Government animal
welfare policy.
ACKNOWLEDGMENTS
We thankChantal Jouanneau, Souhila Ouchelouche, Rémi Piedagnel,
Sandra Onifarasoaniaina, (Institut National de la Santé et de la Recherche
Médicale [INSERM], Unité Mixte de Recherche UMR_S 1155, Paris,
France), Perrine Frere, and Philippe Fontanges (Platerforme d’Imagerie,
Sorbonne Universités, Université Pierre et Marie Curie (UPMC) Univ
Paris 06, Institut Fédératif de Recherche 65, Paris, France) for technical
assistance. We thank Nicolas Sohraindo (Institut Fédératif de Recherche
02, Bichat hospital, Paris, France) for biochemical analysis of urine and
serum sample.
Thisworkwas supportedbygrants fromINSERM,UniversitéPierre et
Marie Curie–Paris 6 (Bonus Qualité Recherche and legs Tessier), Assis-
tance Publique-Hôpitaux de Paris (Délégation à la Recherche Clinique,
Contract CRC06032/P061017), Association pour l’Utilisation du Rein
Artificiel, Agence Nationale de la Recherche (ANR-08-Genopath-018-
02), Délégation Générale à la Santé, through Coordination Theme 1
1052 Journal of the American Society of Nephrology J Am Soc Nephrol 27: 1042–1054, 2016
BASIC RESEARCH www.jasn.org

(Health) of the European Community’s 7th Framework Programme
EUNEFRON (grant agreement no. HEALTH-F2-2007- 201590), Asso-
ciation pour l’Information et la Recherche sur les Maladies Rénales
Génétiques (AIRG-France), and AMGEN Company.
DISCLOSURESNone.
REFERENCES
1. Khoshnoodi J, Pedchenko V, Hudson BG: Mammalian collagen IV.Microsc Res Tech 71: 357–370, 2008
2. Boutaud A, Borza DB, Bondar O, Gunwar S, Netzer KO, Singh N,Ninomiya Y, Sado Y, Noelken ME, Hudson BG: Type IV collagen of theglomerular basement membrane. Evidence that the chain specificity ofnetwork assembly is encoded by the noncollagenous NC1 domains. JBiol Chem 275: 30716–30724, 2000
3. Hudson BG, Tryggvason K, Sundaramoorthy M, Neilson EG: Alport’ssyndrome, Goodpasture’s syndrome, and type IV collagen. N Engl J
Med 348: 2543–2556, 20034. Heino J, HuhtalaM, Käpylä J, JohnsonMS: Evolution of collagen-based
adhesion systems. Int J Biochem Cell Biol 41: 341–348, 20095. Vogel W, Gish GD, Alves F, Pawson T: The discoidin domain receptor
tyrosine kinases are activated by collagen. Mol Cell 1: 13–23, 19976. Leitinger B, Hohenester E: Mammalian collagen receptors. Matrix Biol
26: 146–155, 20077. Gould DB, Phalan FC, Breedveld GJ, van Mil SE, Smith RS, Schimenti
JC, Aguglia U, van der Knaap MS, Heutink P, John SW: Mutations inCol4a1 cause perinatal cerebral hemorrhage and porencephaly. Sci-ence 308: 1167–1171, 2005
8. van der Knaap MS, Smit LM, Barkhof F, Pijnenburg YA, Zweegman S,Niessen HW, Imhof S, Heutink P: Neonatal porencephaly and adult strokerelated to mutations in collagen IV A1. Ann Neurol 59: 504–511, 2006
9. Gould DB, Phalan FC, van Mil SE, Sundberg JP, Vahedi K, Massin P,Bousser MG, Heutink P, Miner JH, Tournier-Lasserve E, John SW: Roleof COL4A1 in small-vessel disease and hemorrhagic stroke. N Engl J
Med 354: 1489–1496, 200610. Lanfranconi S, Markus HS: COL4A1mutations as a monogenic cause of
cerebral small vessel disease: A systematic review. Stroke 41: e513–e518, 2010
11. Yoneda Y, Haginoya K, Kato M, Osaka H, Yokochi K, Arai H, Kakita A,Yamamoto T, Otsuki Y, Shimizu S, Wada T, Koyama N, Mino Y, KondoN, Takahashi S, Hirabayashi S, Takanashi J, Okumura A, Kumagai T,Hirai S, NabetaniM, Saitoh S, Hattori A, YamasakiM, Kumakura A, SugoY, Nishiyama K, Miyatake S, Tsurusaki Y, Doi H, Miyake N, MatsumotoN, Saitsu H: Phenotypic spectrum of COL4A1mutations: Porencephalyto schizencephaly. Ann Neurol 73: 48–57, 2013
12. Coupry I, Sibon I, Mortemousque B, Rouanet F, Mine M, Goizet C:Ophthalmological features associated with COL4A1 mutations. ArchOphthalmol 128: 483–489, 2010
13. Gould DB, Marchant JK, Savinova OV, Smith RS, John SW: Col4a1mutation causes endoplasmic reticulum stress and genetically modifi-able ocular dysgenesis. Hum Mol Genet 16: 798–807, 2007
14. Van Agtmael T, Schlötzer-Schrehardt U, McKie L, Brownstein DG, Lee AW,Cross SH, Sado Y, Mullins JJ, Pöschl E, Jackson IJ: Dominant mutations ofCol4a1 result inbasementmembranedefectswhich lead toanterior segmentdysgenesis and glomerulopathy. HumMol Genet 14: 3161–3168, 2005
15. Favor J, Gloeckner CJ, JanikD, KlemptM,Neuhäuser-KlausA, PretschW,Schmahl W, Quintanilla-Fend L: Type IV procollagen missense mutationsassociated with defects of the eye, vascular stability, the brain, kidneyfunction and embryonic or postnatal viability in themouse,Musmusculus:
An extension of the Col4a1 allelic series and the identification of the firsttwo Col4a2 mutant alleles. Genetics 175: 725–736, 2007
16. Plaisier E, Gribouval O, Alamowitch S, Mougenot B, Prost C, VerpontMC, Marro B, Desmettre T, Cohen SY, Roullet E, DraconM, Fardeau M,Van Agtmael T, Kerjaschki D, Antignac C, Ronco P: COL4A1 mutationsand hereditary angiopathy, nephropathy, aneurysms, and musclecramps. N Engl J Med 357: 2687–2695, 2007
17. Plaisier E, Alamowitch S, Gribouval O, Mougenot B, Gaudric A,Antignac C, Roullet E, Ronco P: Autosomal-dominant familial hema-turia with retinal arteriolar tortuosity and contractures: A novel syn-drome. Kidney Int 67: 2354–2360, 2005
18. Plaisier E, Chen Z, Gekeler F, Benhassine S, Dahan K, Marro B,Alamowitch S, Paques M, Ronco P: Novel COL4A1 mutations associ-ated with HANAC syndrome: A role for the triple helical CB3[IV] do-main. Am J Med Genet A 152A: 2550–2555, 2010
19. Alamowitch S, Plaisier E, Favrole P, Prost C, Chen Z, Van Agtmael T,Marro B, Ronco P: Cerebrovascular disease related to COL4A1 muta-tions in HANAC syndrome. Neurology 73: 1873–1882, 2009
20. Kuo DS, Labelle-Dumais C, Mao M, Jeanne M, Kauffman WB, Allen J,Favor J, Gould DB: Allelic heterogeneity contributes to variability inocular dysgenesis, myopathy and brain malformations caused byCol4a1 and Col4a2 mutations. Hum Mol Genet 23: 1709–1722, 2014
21. Jais JP, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, Renieri A,Weber M, Gross O, Netzer KO, Flinter F, Pirson Y, Verellen C,Wieslander J, PerssonU, Tryggvason K,Martin P, Hertz JM, Schröder C,Sanak M, Krejcova S, Carvalho MF, Saus J, Antignac C, Smeets H,Gubler MC: X-linked Alport syndrome: Natural history in 195 familiesand genotype- phenotype correlations in males. J Am Soc Nephrol 11:649–657, 2000
22. Bekheirnia MR, Reed B, Gregory MC, McFann K, Shamshirsaz AA,Masoumi A, Schrier RW: Genotype-phenotype correlation in X-linkedAlport syndrome. J Am Soc Nephrol 21: 876–883, 2010
23. Storey H, Savige J, Sivakumar V, Abbs S, Flinter FA: COL4A3/COL4A4mutations and features in individuals with autosomal recessive Alportsyndrome. J Am Soc Nephrol 24: 1945–1954, 2013
24. Miner JH: Developmental biology of glomerular basement membranecomponents. Curr Opin Nephrol Hypertens 7: 13–19, 1998
25. Kreidberg JA, Donovan MJ, Goldstein SL, Rennke H, Shepherd K,Jones RC, Jaenisch R: Alpha 3 beta 1 integrin has a crucial role in kidneyand lung organogenesis. Development 122: 3537–3547, 1996
26. Kanasaki K, Kanda Y, Palmsten K, Tanjore H, Lee SB, Lebleu VS,Gattone VH Jr, Kalluri R: Integrin beta1-mediated matrix assembly andsignaling are critical for the normal development and function of thekidney glomerulus. Dev Biol 313: 584–593, 2008
27. Korhonen M, Ylänne J, Laitinen L, Virtanen I: The a 1-a 6 subunits ofintegrins are characteristically expressed in distinct segments of de-veloping and adult human nephron. J Cell Biol 111: 1245–1254, 1990
28. Zent R, Bush KT, PohlML,Quaranta V, KoshikawaN,WangZ, KreidbergJA, Sakurai H, Stuart RO, Nigám SK: Involvement of laminin bindingintegrins and laminin-5 in branchingmorphogenesis of the ureteric budduring kidney development. Dev Biol 238: 289–302, 2001
29. Delwel GO, de Melker AA, Hogervorst F, Jaspars LH, Fles DL, KuikmanI, Lindblom A, Paulsson M, Timpl R, Sonnenberg A: Distinct and over-lapping ligand specificities of the alpha 3A beta 1 and alpha 6A beta 1integrins: Recognition of laminin isoforms. Mol Biol Cell 5: 203–215,1994
30. Miner JH, Sanes JR: Collagen IV alpha 3, alpha 4, and alpha 5 chains inrodent basal laminae: Sequence, distribution, association with lam-inins, and developmental switches. J Cell Biol 127: 879–891, 1994
31. Lennerz JK, Spence DC, Iskandar SS, Dehner LP, Liapis H: Glomer-ulocystic kidney: One hundred-year perspective. Arch Pathol Lab Med
134: 583–605, 201032. Smeets B, Uhlig S, Fuss A,Mooren F, Wetzels JF, Floege J, Moeller MJ:
Tracing the origin of glomerular extracapillary lesions from parietalepithelial cells. J Am Soc Nephrol 20: 2604–2615, 2009
J Am Soc Nephrol 27: 1042–1054, 2016 Renal Defects in HANAC Col4a1 Mice 1053
www.jasn.org BASIC RESEARCH

33. Smeets B, Kuppe C, Sicking EM, Fuss A, Jirak P, van Kuppevelt TH,Endlich K, Wetzels JF, Gröne HJ, Floege J, Moeller MJ: Parietal epi-thelial cells participate in the formation of sclerotic lesions in focalsegmental glomerulosclerosis. J AmSocNephrol 22: 1262–1274, 2011
34. Shankland SJ, Smeets B, Pippin JW,Moeller MJ: The emergence of theglomerular parietal epithelial cell. Nat Rev Nephrol 10: 158–173, 2014
35. Shrivastava A, Radziejewski C, Campbell E, Kovac L, McGlynn M, RyanTE, Davis S, Goldfarb MP, Glass DJ, Lemke G, Yancopoulos GD: Anorphan receptor tyrosine kinase family whose members serve as non-integrin collagen receptors. Mol Cell 1: 25–34, 1997
36. Fu HL, Valiathan RR, Arkwright R, Sohail A, Mihai C, Kumarasiri M,Mahasenan KV, Mobashery S, Huang P, Agarwal G, Fridman R: Discoidindomain receptors: Unique receptor tyrosine kinases in collagen-mediatedsignaling. J Biol Chem 288: 7430–7437, 2013
37. KerrochM, Guerrot D, Vandermeersch S, Placier S, Mesnard L, JouanneauC, Rondeau E, Ronco P, Boffa JJ, Chatziantoniou C, Dussaule JC: Geneticinhibition of discoidin domain receptor 1 protects mice against crescenticglomerulonephritis. FASEB J 26: 4079–4091, 2012
38. Guerrot D, KerrochM, Placier S, Vandermeersch S, Trivin C, Mael-AininM, Chatziantoniou C, Dussaule JC: Discoidin domain receptor 1 is amajor mediator of inflammation and fibrosis in obstructive nephropa-thy. Am J Pathol 179: 83–91, 2011
39. Teixeira VP, Blattner SM, Li M, Anders HJ, Cohen CD, Edenhofer I,Calvaresi N, Merkle M, Rastaldi MP, Kretzler M: Functional con-sequences of integrin-linked kinase activation in podocyte damage.Kidney Int 67: 514–523, 2005
40. El-Aouni C, Herbach N, Blattner SM, Henger A, Rastaldi MP, Jarad G,Miner JH, Moeller MJ, St-Arnaud R, Dedhar S, Holzman LB, Wanke R,Kretzler M: Podocyte-specific deletion of integrin-linked kinase resultsin severe glomerular basement membrane alterations and progressiveglomerulosclerosis. J Am Soc Nephrol 17: 1334–1344, 2006
41. Dai C, Stolz DB, Bastacky SI, St-Arnaud R, Wu C, Dedhar S, Liu Y: Es-sential role of integrin-linked kinase in podocyte biology: Bridging theintegrin and slit diaphragm signaling. J Am Soc Nephrol 17: 2164–2175, 2006
42. Wickström SA, Lange A, Montanez E, Fässler R: The ILK/PINCH/parvincomplex: The kinase is dead, long live the pseudokinase! EMBO J 29:281–291, 2010
43. Joshi MB, Ivanov D, Philippova M, Erne P, Resink TJ: Integrin-linkedkinase is an essential mediator for T-cadherin-dependent signaling viaAkt and GSK3beta in endothelial cells. FASEB J 21: 3083–3095, 2007
44. Miwa N, Furuse M, Tsukita S, Niikawa N, Nakamura Y, Furukawa Y:Involvement of claudin-1 in the beta-catenin/Tcf signaling pathway andits frequent upregulation in human colorectal cancers. Oncol Res 12:469–476, 2001
45. Wielenga VJ, Smits R, Korinek V, Smit L, KielmanM, FoddeR, CleversH,Pals ST: Expression of CD44 in Apc and Tcf mutant mice implies reg-ulation by the WNT pathway. Am J Pathol 154: 515–523, 1999
46. Novak A, Hsu SC, Leung-Hagesteijn C, Radeva G, Papkoff J,Montesano R, Roskelley C, Grosschedl R, Dedhar S: Cell adhesion andthe integrin-linked kinase regulate the LEF-1 and beta-catenin signal-ing pathways. Proc Natl Acad Sci U S A 95: 4374–4379, 1998
47. Somasiri A, Howarth A, Goswami D, Dedhar S, Roskelley CD: Over-expression of the integrin-linked kinase mesenchymally transformsmammary epithelial cells. J Cell Sci 114: 1125–1136, 2001
48. Gil D, Ciołczyk-Wierzbicka D, Duli�nska-Litewka J, Zwawa K, McCubreyJA, Laidler P: The mechanism of contribution of integrin linked kinase(ILK) to epithelial-mesenchymal transition (EMT).AdvEnzymeRegul51:195–207, 2011
49. Troussard AA, Costello P, Yoganathan TN, Kumagai S, Roskelley CD,Dedhar S: The integrin linked kinase (ILK) induces an invasive pheno-type via AP-1 transcription factor-dependent upregulation of matrixmetalloproteinase 9 (MMP-9). Oncogene 19: 5444–5452, 2000
50. Li Y, Yang J, Dai C, Wu C, Liu Y: Role for integrin-linked kinase in me-diating tubular epithelial to mesenchymal transition and renal in-terstitial fibrogenesis. J Clin Invest 112: 503–516, 2003
51. Zhang YH, Huang BL, Eastman K,McCabe LL,MacLennan NK,McCabeER: Mouth cell collection device for newborn mice. Mol Genet Metab89: 164–167, 2006
52. Regele HM, Fillipovic E, Langer B, Poczewki H, Kraxberger I, Bittner RE,Kerjaschki D: Glomerular expression of dystroglycans is reduced inminimal change nephrosis but not in focal segmental glomerulo-sclerosis. J Am Soc Nephrol 11: 403–412, 2000
53. Mook OR, Van Overbeek C, Ackema EG, Van Maldegem F, FrederiksWM: In situ localization of gelatinolytic activity in the extracellular ma-trix of metastases of colon cancer in rat liver using quenched fluoro-genic DQ-gelatin. J Histochem Cytochem 51: 821–829, 2003
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2014121217/-/DCSupplemental.
1054 Journal of the American Society of Nephrology J Am Soc Nephrol 27: 1042–1054, 2016
BASIC RESEARCH www.jasn.org


















