Exploration des Hypertrophies Ventriculaires Gauches...
80
Exploration des Hypertrophies Ventriculaires Gauches en IRM Cardiaque Jérôme Caudron¹, Hend Belhiba¹, Jeannette Fares², David Bertrand¹ et Jean-Nicolas Dacher¹ 1- Service de Radiologie Centrale – Hôpital Charles Nicolle – Rouen 2- Service de Cardiologie – Hôpital Charles Nicolle – Rouen Contact : [email protected]
Transcript of Exploration des Hypertrophies Ventriculaires Gauches...

Exploration des Hypertrophies Ventriculaires Gauches en IRM Cardiaque
Jérôme Caudron¹, Hend Belhiba¹, Jeannette Fares², David Bertrand¹et Jean-Nicolas Dacher¹
1- Service de Radiologie Centrale – Hôpital Charles Nicolle – Rouen
2- Service de Cardiologie – Hôpital Charles Nicolle – Rouen
Contact : [email protected]

Liste des abréviations utilisées
HVG: Hypertrophie Ventriculaire GaucheVG: Ventricule GaucheVD: Ventricule DroitFEVG: Fraction d’Ejection Ventriculaire GaucheOG: Oreillette GaucheOD: Oreillette DroiteSIV: Septum Inter-VentriculaireCMH: Cardiomyopathie HypertrophiqueECG: ElectrocardiogrammeETT: Echocardiographie Trans-ThoraciqueRA: Rétrécissement AortiqueTi: Temps d’inversionESV: Extra Systoles VentriculairesTVNS: Tachycardie Ventriculaire Non Soutenue

PlanPrincipales causes d’HVGDéfinition de l’HVGExploration des HVG
ECG et ETTIRM
Revue iconographiqueCMHAmylose cardiaqueRétrécissements aortiquesHVG physiologique du sportifSarcoïdoseMaladie de FabryHTATumeurs cardiaques localisées au VGPièges diagnostiques
ConclusionBibliographie
Cliquez sur le chapitre de votre choix pour y
accéder
Cliquez sur
pour revenir au menu

Principales causes d’HVG
Cause primitive:Cardiomyopathies hypertrophiques (CMH)
Causes secondaires:Augmentation de la post-charge
HTARétrécissements aortiques (RA)Coarctation aortique
Infiltration myocardiqueAmylose cardiaqueGranulomatoses : SarcoïdoseMaladie de FabryTumeurs cardiaques
HVG physiologique du sportif
Le diagnostic différentiel entre ces différentes étiologies est primordial et parfois difficile car elles peuvent être intriquées: HTA et RA, CMH et HTA, Amylose et RA, HTA et
sport, CMH et sport…

Définition de l’HVG
Epaississement pariétal du VG > 11 mmMesuré en diastole.Sur des séquences SSFP (TRUFISP, FIESTA…):
Coupes petit axe au niveau basal et médian.Coupes LAVG et 4 cavités au niveau de l’apex (en prenant garde à être bien perpendiculaire à la paroi du VG pour ne pas surestimer l’HVG).
Pièges: faux tendons du côté du VD et trabéculations du côté du VG, parfois difficiles à distinguer de la paroi propre.Topographie variable selon l’étiologie: diffus (ex: HTA) ou localisé (ex: CMH).
Augmentation de la masse VGL’IRM est la technique de référence pour évaluer la masse VG (1).L’augmentation de la masse VG est définie dans notre centre par une masse VG:
Chez les hommes > 238g en valeur absolue, > 113g/m² en valeur indexée.Chez les femmes > 175g en valeur absolue, > 95g/m² en valeur indexée.
La masse VG peut être normale en cas d’hypertrophie focalisée, par exemple en cas de CMH où elle est normale dans 20% des cas

Définition de l’HVG
Plusieurs types de géométrie VG ont été définies en ETT dans le cadre des HVG secondaires à une augmentation de la post charge (HTA et RA+++).
EPI: Epaisseur pariétale postérieure indexée au diamètre télédiastolique VGMVGI: masse VG indexée à la surface corporelle

Définition de l’HVG
On peut donc concevoir la définition de l’HVG de 2 façons:
Augmentation de la masse VG: définition habituelle et utilisée dans toutes les grandes études explorant l’HTA notamment.
Epaississement anormal du VG: une HVG peut en effet être focalisée sans augmenter la masse VG, comme par exemple en cas de CMH ou de tumeur cardiaque.
Dans ce poster ont été considérées comme causes d’HVG toutes les pathologies les plus courantes susceptibles d’entrainer une augmentation de l’épaisseur et/ou de la masse ventriculaire gauche, que l’atteinte soit focale ou diffuse.
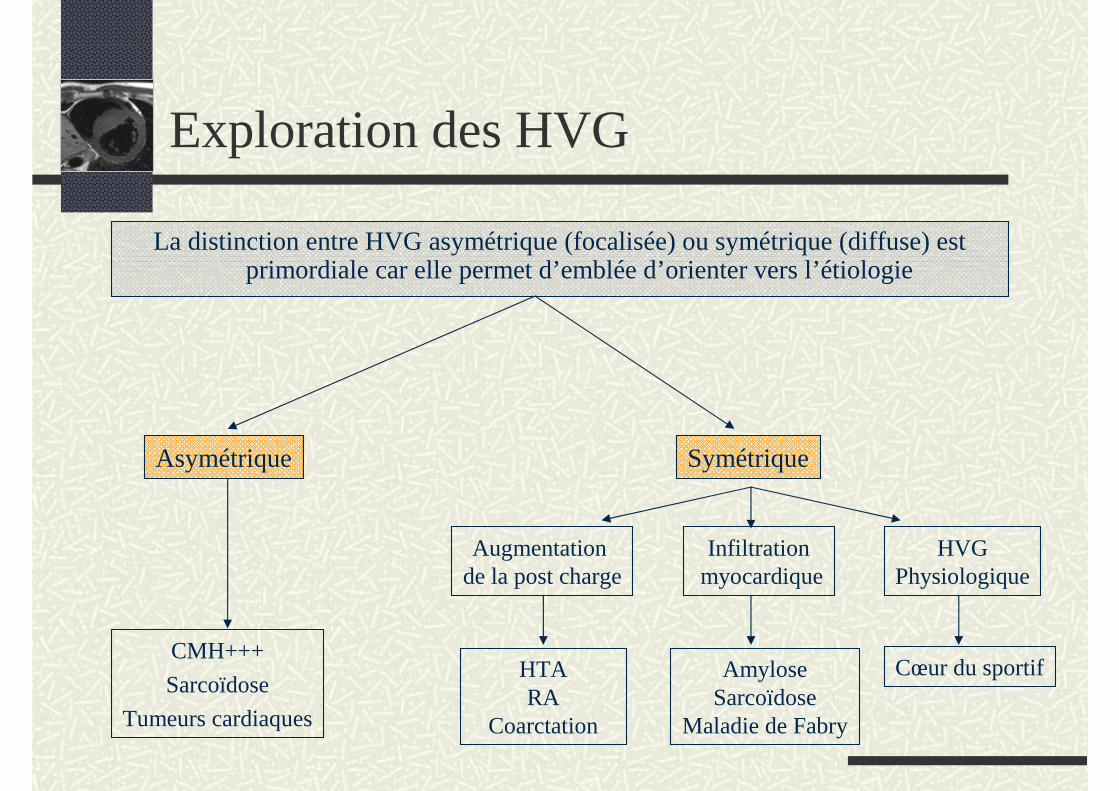
Exploration des HVG
La distinction entre HVG asymétrique (focalisée) ou symétrique (diffuse) est primordiale car elle permet d’emblée d’orienter vers l’étiologie
Asymétrique
CMH+++
Sarcoïdose
Tumeurs cardiaques
Symétrique
Augmentation de la post charge
Infiltrationmyocardique
HVGPhysiologique
HTARA
Coarctation
AmyloseSarcoïdose
Maladie de Fabry
Cœur du sportif
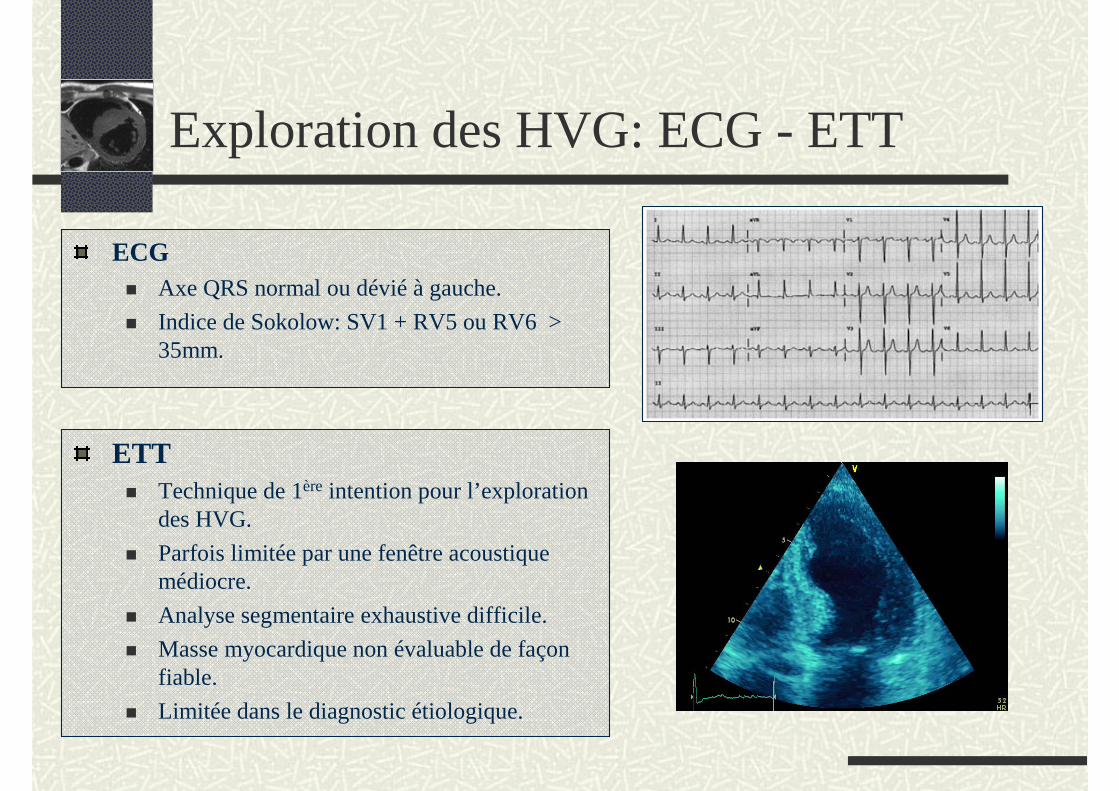
Exploration des HVG: ECG - ETT
ECG Axe QRS normal ou dévié à gauche.
Indice de Sokolow: SV1 + RV5 ou RV6 > 35mm.
ETTTechnique de 1ère intention pour l’exploration des HVG.
Parfois limitée par une fenêtre acoustique médiocre.
Analyse segmentaire exhaustive difficile.
Masse myocardique non évaluable de façon fiable.
Limitée dans le diagnostic étiologique.

Exploration des HVG: IRM
L’IRM cardiaque possède un intérêt certain dans l’exploration des HVG:
Etude exhaustive de l’ensemble du VG et des autres structures anatomiques cardiaques.Localisation et quantification précise des zones hypertrophiées.Masse myocardique fiable et reproductible (1).Examen de référence pour l’étude de la FEVG et des volumes VG.Etude de la perfusion myocardique.Fonction diastolique évaluable.Séquences de rehaussement tardif (RT) très utiles au diagnostic étiologique (2).
L’IRM cardiaque est donc un examen clef dans le bilan étiologique et pronostique des HVG, en complément des explorations habituelles

Exploration des HVG: IRM
Technique d’examen dans notre centre:IRM Siemens Symphony 1,5T, antenne cardiaque dédiéeInstallation du patient et vérification de la bonne qualité du signal ECGSéquences de repérage morphologique non synchronisées dans les 3 plans permettant de définir les plans de référenceSéquences Ciné SSFP en 4 cavités, LAVG et petit axe de la base à l’apex pour l’étude de la cinétique segmentaire, de l’épaisseur et de la masse myocardique, des volumes VG et le la FEVGSéquences morphologiques T2 STIR FSE dans les 3 plans en fonction du contexte clinique.Perfusion premier passage en imagerie rapide, avec injection d’emblée de 0,3 mmol/Kg de Gadolinium à fort débit (6cc/s)Séquences Ciné SSFP LVOT 1 et 2 puis séquence sur la valve aortique, très utiles pour évaluer le bourrelet sous aortique dans les CMH et éliminer une sténose aortiqueSéquences en contraste de phase petit axe au niveau de l’entonnoir mitral pour l’étude de la fonction diastoliqueSéquences de rehaussement tardif dans les 3 plans après calcul du temps d’inversion optimal sur la séquence de Ti scoutingSéquences PSIR en complément du RT si besoin

Hypertrophie anormale primitive du VG associée à une désorganisation cellulaire, prédominant le plus souvent au niveau septal, avec développement d’une fibrose intercellulaire et donc augmentation anormale du tissu conjonctif de soutien (collagène) (3).
CMH: Généralités
Myocarde normal CMH
Prévalence phénotypique estimée à 0,2%.Origine génétique avec une majorité de formes familiales et mutations de gènes codant pour des protéines du sarcomère (actine, myosine, troponine…).Transmission autosomique dominante à pénétrance variable, avec plus de 150 mutations décrites à ce jour.> 50% des formes sont asymptomatiques, donc la prévalence génotypique est probablement nettement supérieure à la prévalence phénotypique.

CMH: Généralités
Conséquences de l’HVG :Anomalies de la fonction diastolique associant troubles de la relaxation et troubles de la compliance, conséquences de l’hypertrophie pariétale importante.Obstruction sur la voie d’éjection du VG dans 25% des cas, habituellement au niveau de la chambre de chasse, phénomène entretenant l’HVG.Ischémie myocardique avec diminution de la réserve coronaire car densitécapillaire inappropriée par rapport à l’HVG.Arythmies avec risque de mort subite.
Options thérapeutiques :Traitement médical : bêtabloquants et inhibiteurs calciques.Traitement de l’obstruction intra-VG : 3 options
Stimulation double chambre.Alcoolisation de la première artère septale (4) avec dans ce cas un bilan IRM pré et post intervention indispensable. Chirurgie avec myectomie.

CMH: Généralités
Le risque annuel de mort subite en cas de CMH est de:1% pour les adultes et 2 à 4 % pour les enfants et les adolescents.
Le plus souvent sur troubles du rythme ventriculaire.
Facteurs de risque classiques de mort subite chez les patients ayant une CMH:
ATCD de mort subite récupérée ou de TVS.
ATCD familiaux de multiples morts subites.
ATCD de syncope inexpliquée.
Epaississement pariétal myocardique >30 mm.
Elévation anormale de la TA à l’effort.
TVNS sur Holter ECG des 24h.
Nous reviendrons plus loin sur le rôle potentiel de l’IRM pour identifier les patients à risque de mort subite en IRM.

CMH et IRM: Morphologie
Epaisseur pariétale maximale anormale:≥ 15 mm dans les formes sporadiques avec absence de dilatation du VG ou de
pathologie pouvant expliquer l’HVG, mais dans la majeure partie des cas elle est déjà > 20 mm et dans 12% des cas > 30 mm (5).≥ 13 mm dans les formes familiales et ≥ 16 mm chez le sportif (6, 7).L’IRM permet de détecter des zones hypertrophiées non détectées par l’ETT et est plus précise pour la quantification de l’HVG notamment au niveau antéro-latéro-basal et pour les hypertrophies importantes ≥ 30 mm (8).
Masse VG:La masse VG est normale chez environ 20% des patients présentant un phénotype de CMH définis par l’ETT (9): son augmentation n’est donc pas un critère indispensable au diagnostic positif de CMH.
La corrélation entre masse VG et épaisseur pariétale maximale est mauvaise mais la masse VG est corrélée à l’importance du RT et à l’existence d’une obstruction intra-VG (9).
L’augmentation de la masse VG est plus sensible (mais moins spécifique) qu’une épaisseur pariétale maximale > 30 mm pour prédire le risque de décès (9).
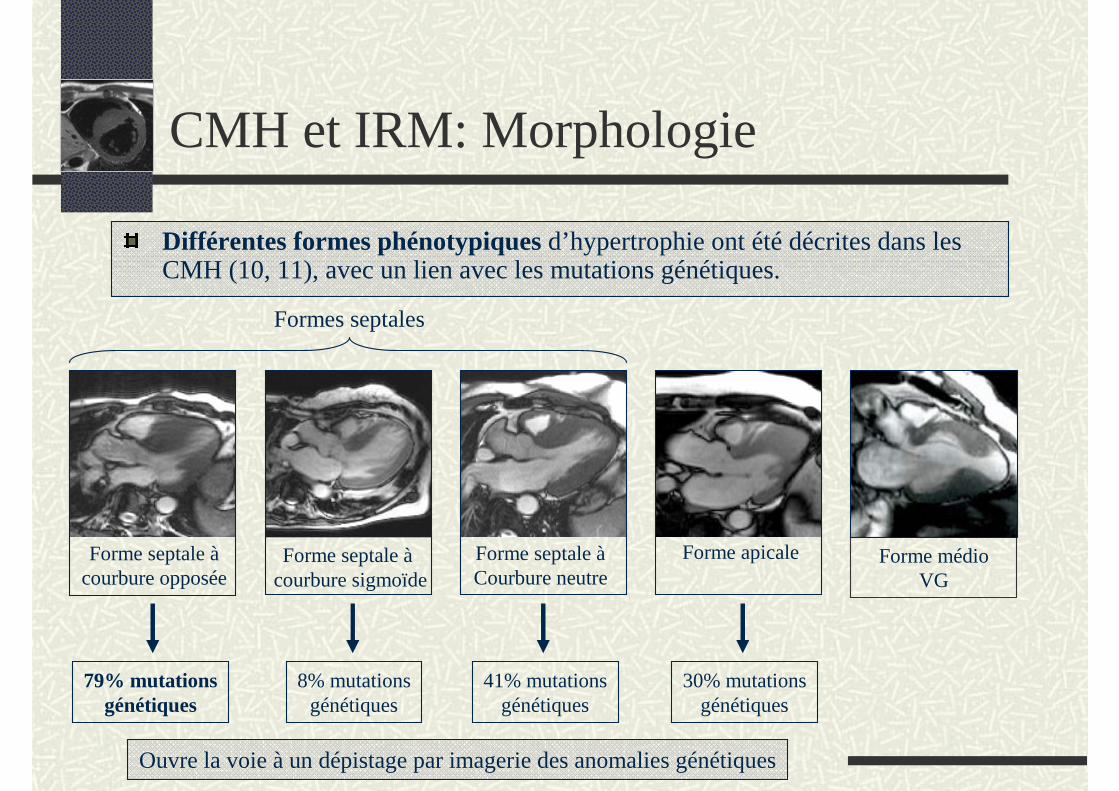
CMH et IRM: Morphologie
Différentes formes phénotypiques d’hypertrophie ont été décrites dans les CMH (10, 11), avec un lien avec les mutations génétiques.
Ouvre la voie à un dépistage par imagerie des anomalies génétiques
Forme septale àcourbure opposée
Forme septale àcourbure sigmoïde
Forme septale àCourbure neutre
Forme apicale
79% mutationsgénétiques
8% mutationsgénétiques
41% mutationsgénétiques
30% mutationsgénétiques
Formes septales
Forme médio VG
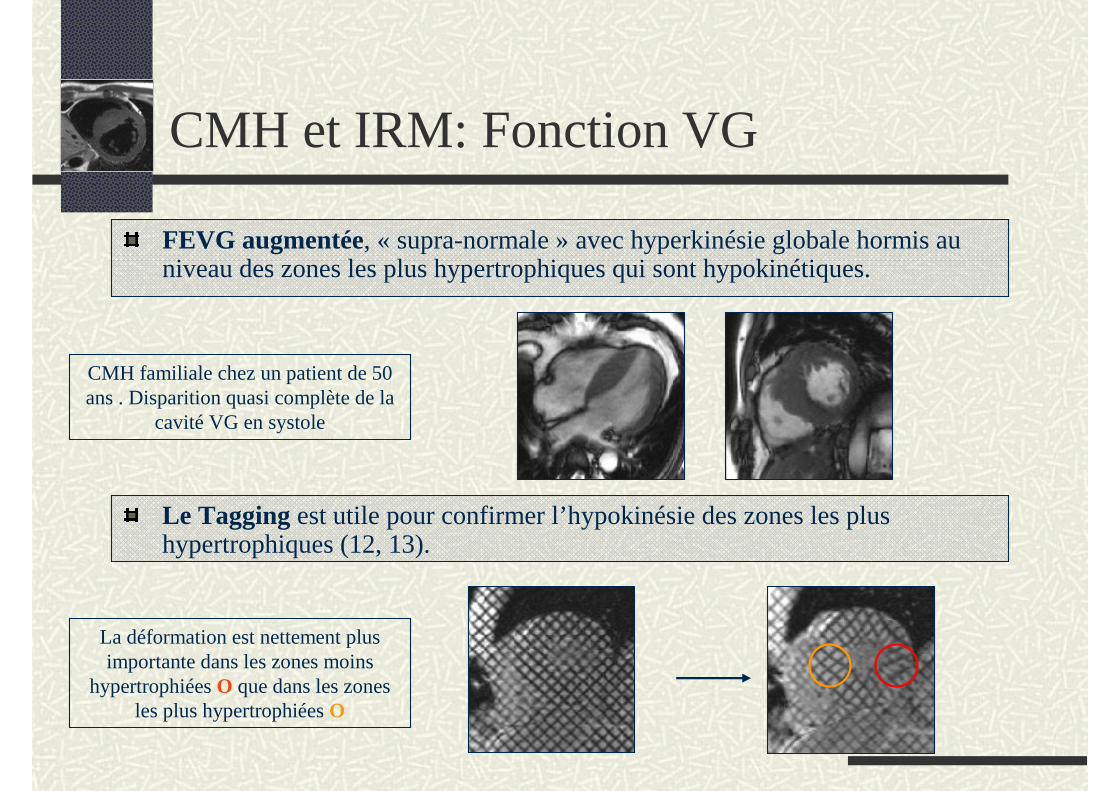
CMH et IRM: Fonction VG
FEVG augmentée, « supra-normale » avec hyperkinésie globale hormis au niveau des zones les plus hypertrophiques qui sont hypokinétiques.
Le Tagging est utile pour confirmer l’hypokinésie des zones les plus hypertrophiques (12, 13).
La déformation est nettement plus importante dans les zones moins
hypertrophiées O que dans les zones les plus hypertrophiées O
CMH familiale chez un patient de 50 ans . Disparition quasi complète de la
cavité VG en systole
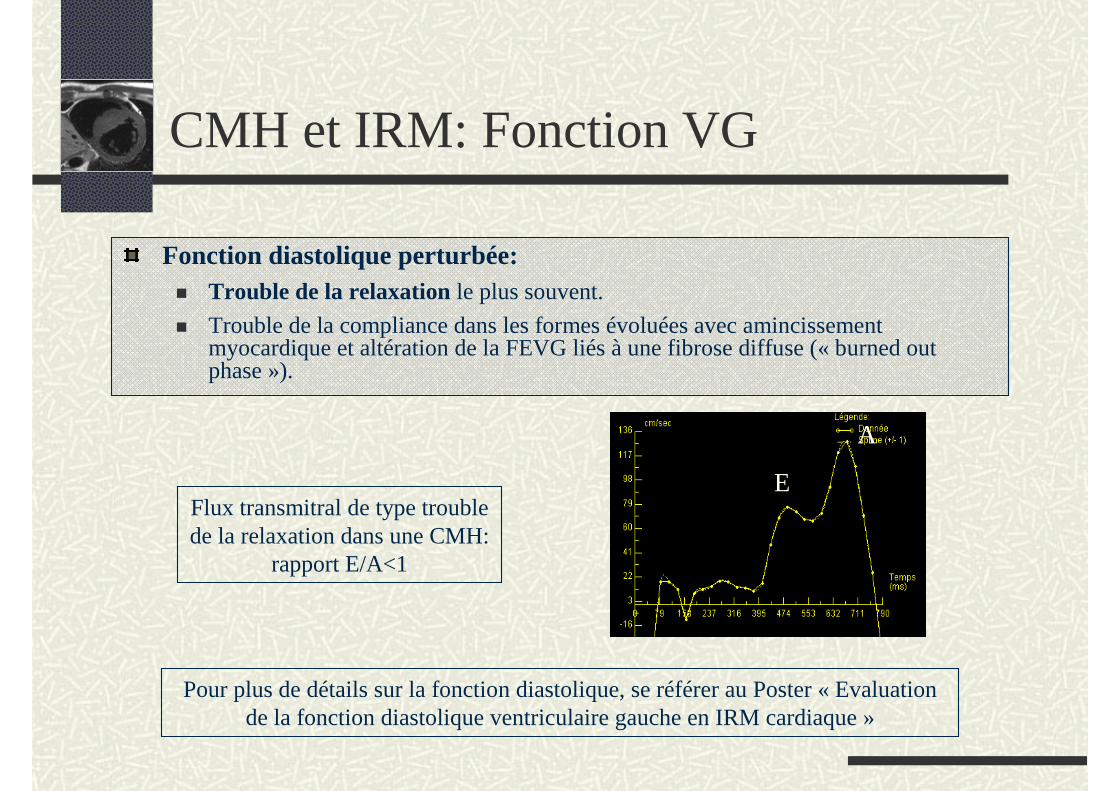
CMH et IRM: Fonction VG
Fonction diastolique perturbée:Trouble de la relaxation le plus souvent.
Trouble de la compliance dans les formes évoluées avec amincissement myocardique et altération de la FEVG liés à une fibrose diffuse (« burned out phase »).
Flux transmitral de type trouble de la relaxation dans une CMH:
rapport E/A<1
Pour plus de détails sur la fonction diastolique, se référer au Poster « Evaluation de la fonction diastolique ventriculaire gauche en IRM cardiaque »
E
A
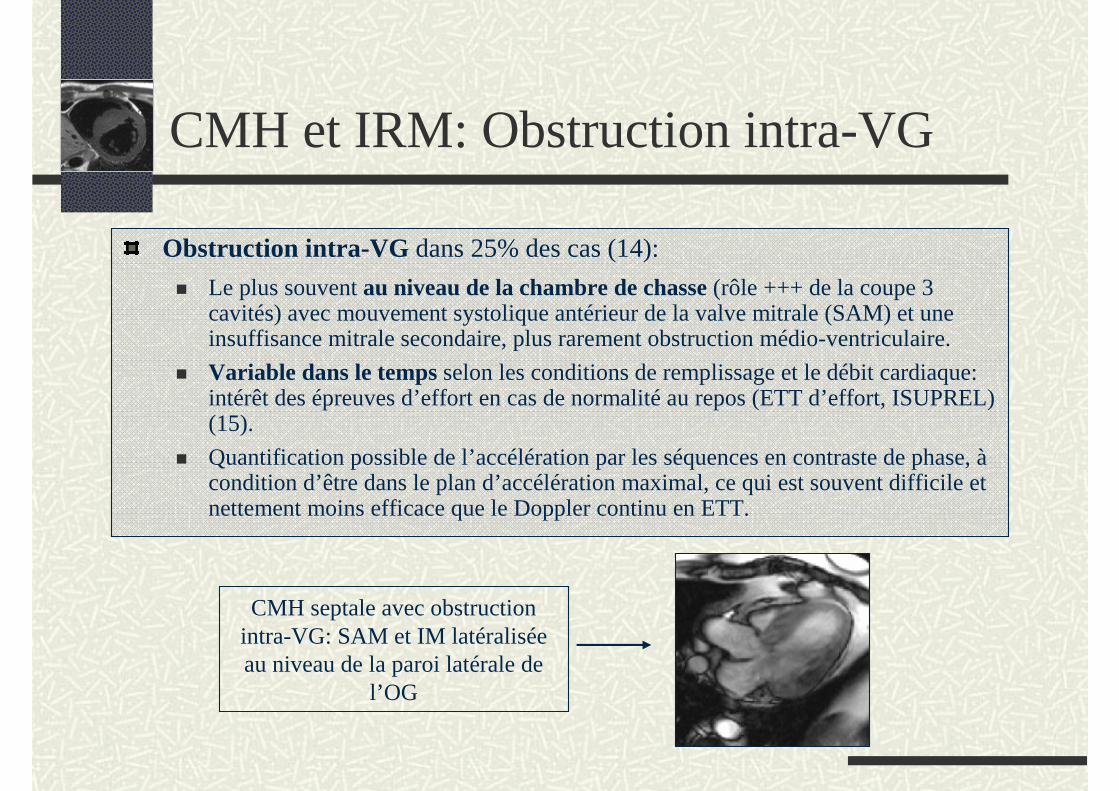
CMH et IRM: Obstruction intra-VG
Obstruction intra-VG dans 25% des cas (14):
Le plus souvent au niveau de la chambre de chasse (rôle +++ de la coupe 3 cavités) avec mouvement systolique antérieur de la valve mitrale (SAM) et une insuffisance mitrale secondaire, plus rarement obstruction médio-ventriculaire.
Variable dans le temps selon les conditions de remplissage et le débit cardiaque: intérêt des épreuves d’effort en cas de normalité au repos (ETT d’effort, ISUPREL) (15).
Quantification possible de l’accélération par les séquences en contraste de phase, àcondition d’être dans le plan d’accélération maximal, ce qui est souvent difficile et nettement moins efficace que le Doppler continu en ETT.
CMH septale avec obstruction intra-VG: SAM et IM latéralisée au niveau de la paroi latérale de
l’OG

CMH et IRM: Perfusion
Perfusion de repos:La perfusion 1er passage au repos est le plus souvent normale mais possible hypoperfusion notamment sous endocardique (16).
Perfusion sous stress (17, 18):Les anomalies de perfusion sont plus souvent constatées en cas d’épreuve pharmacologique (Adénosine, Dipyridamole) avec une diminution de la perfusion sous endocardique liée à une dysfonction de la microcirculation coronaire (19) expliquant les possibles douleurs angineuses malgré des artères coronaires angiographiquement normales.
Ces anomalies sont corrélées à l’importance de l’hypertrophie et aux évènements indésirables type mort subite ou altération de la FEVG (20, 21).
Par ailleurs les zones qui ont un important RT ont une baisse plus importante de la perfusion myocardique que les zones sans RT (22, 23) suggérant un lien direct entre les anomalies de la microcirculation et la fibrose myocardique.
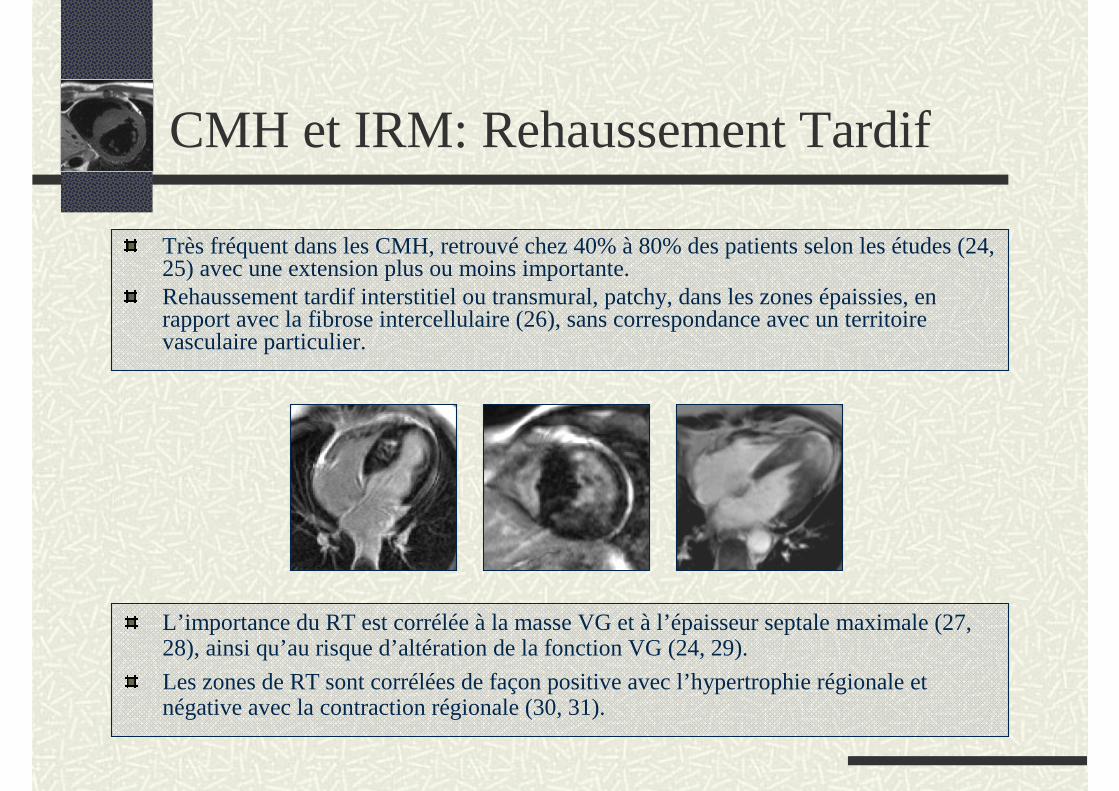
Très fréquent dans les CMH, retrouvé chez 40% à 80% des patients selon les études (24, 25) avec une extension plus ou moins importante.Rehaussement tardif interstitiel ou transmural, patchy, dans les zones épaissies, en rapport avec la fibrose intercellulaire (26), sans correspondance avec un territoire vasculaire particulier.
CMH et IRM: Rehaussement Tardif
L’importance du RT est corrélée à la masse VG et à l’épaisseur septale maximale (27, 28), ainsi qu’au risque d’altération de la fonction VG (24, 29).
Les zones de RT sont corrélées de façon positive avec l’hypertrophie régionale et négative avec la contraction régionale (30, 31).

CMH et IRM: Rehaussement Tardif
Le rehaussement tardif en IRM : un nouveau facteur de risque de mort subite?
Intérêt majeur car identification possible du substrat arythmogène: les zones de RT correspondent aux zones de désorganisation architecturales les plus marquées avec fibrose et infiltration collagénique importante (26, 32).
Ce qui est démontré (24, 25): la présence d’un rehaussement tardif est corrélée à la présence de facteurs de risque de mort subite notamment TVNS, ESV et Doublets d’ESV sur un enregistrement Holter des 24 h.
Ce qui doit être démontré: La démonstration de façon prospective que l’incidence des morts subites est plus élevée chez les patients ayant un RT reste à démontrer même si elle est très probable au vu des différents travaux publiés (33).

CMH et IRM: Contrôle post alcoolisation
L’IRM joue un rôle important dans l’évaluation des patients ayant bénéficié d’une alcoolisation septale (34-36).
L’alcoolisation entraîne un infarctus localisé dans le territoire de la première artère septale.
L’IRM permet de quantifier l’étendue de l’infarctus (séquences de RT), d’évaluer la diminution de la taille du bourrelet et la disparition du SAM (séquences SSFP), et de suivre la masse VG qui diminue d’une part via la disparition du bourrelet mais également via la diminution de la post charge (37).
Patiente de 74 ans – CMH obstructive symptomatique
traitée par alcoolisation septale
Séquence SSFP 3 cavités avant alcoolisation:
bourrelet septal obstructif avec SAM et IM
Contrôle 3 mois après alcoolisation: régression du bourrelet septal, du
SAM et de l’IM
Rehaussement tardif trans mural avec no reflow dans le
territoire de la 1ère septale

CMH et IRM: Points Clefs
Au niveau morphologiqueEpaisseur pariétale VG anormale*, prédominant le plus souvent au niveau septal.Masse VG souvent augmentée mais normale dans 20% des cas.
Au niveau fonctionnelFEVG augmentée*, « supra-normale » avec hyperkinésie globale hormis au niveau des zones les plus hypertrophiques qui sont hypokinétiques.Fonction diastolique perturbée*: trouble de la relaxation le plus souvent, trouble de la compliance dans les formes évoluées avec amincissement myocardique et altération de la FEVG liés à une fibrose diffuse.Obstruction intra-ventriculaire (25% des cas):
Le plus souvent au niveau de la chambre de chasse avec SAM et insuffisance mitrale secondaire, plus rarement obstruction médio-ventriculaire.Variable dans le temps selon les conditions de remplissage et le débit cardiaque: intérêt des épreuves d’effort (ETT d’effort, ISUPREL).
Perfusion premier passage le plus souvent normale mais possible hypoperfusion notamment sous endocardique, surtout en cas d’épreuve pharmacologique.
Rehaussement tardifRehaussement tardif interstitiel ou transmural, patchy, dans les zones épaissies, en rapport avec la fibrose intercellulaire
* Signes constants

Amylose Cardiaque: Généralités
HVG en rapport avec des dépôts extra cellulaires de substance amyloïde,prédominant au niveau sous-endocardique.
Plusieurs types d’amylose (38):
Amylose de type primitive (AL), le plus souvent secondaire à un myélome multiple ou à une autre gammapathie monoclonale, avec dépôts de chaînes légères dans tous les tissus.
Amylose de type secondaire (AA), secondaire à des pathologies inflammatoires chroniques (polyarthrite rhumatoïde, BK…).
(Amylose systémique héréditaire et amylose systémique sénile).
Atteinte cardiaque nettement plus fréquente en cas d’amylose AL, tandis que l’atteinte rénale domine le tableau des amyloses AA.
Pronostic très sombre (38).

Amylose Cardiaque: Généralités
Clinique:
Contexte parfois évocateur (myélome multiple, gammapathie monoclonale, autre localisation amyloïde).
Tableau clinique variable: cardiomyopathie restrictive, insuffisance cardiaque systolique, hypotension orthostatique (infiltration du système nerveux autonome et/ou des vaisseaux) et troubles de la conduction et/ou du rythme.
Diagnostic habituel par ECG (microvoltage ou pseudo infarctus avec ondes Q) et ETT montrant une HVG importante avec myocarde hyperéchogène d’aspect « scintillant » (39).
Gold standard : Biopsie endomyocardique avec examen anatomo-pathologique (coloration rouge Congo) montrant une infiltration extra cellulaire par une protéine fibrillaire, prédominant au niveau sous endocardique, associée à une fibrose endomyocardique. En cas d’amylose systémique, des biopsies moins invasives peuvent être utilisées (muqueuse rectale, graisse sous cutanée abdominale) (40).

Amylose et IRM: Morphologie
HVG concentrique avec une cavité ventriculaire gauche de petite taille.
Augmentation du temps de relaxation T1 et T2 (41, 42).
Dilatation bi auriculaire et épaississement de la paroi libre de l’OD (> 6mm) et du septum inter-auriculaire (> 6mm) (43, 44).
Epaississement de la paroi libre du VD, diffuse (alors que plutôt apicale dans les CMH) (43, 45)
Epanchements péricardiques et pleuraux fréquents.
Patient de 60 ans. Amylose cardiaque de type AL. • HVG concentrique (septum à 20 mm, masse myocardique à 148 g/m²), sans dilatation du VG.• Epaississement de la paroi libre de l’OD et du septum inter-auriculaire avec dilatation biauriculaire.• Epanchement péricardique circonférentiel non compressif
Amylose et IRM: Fonction VG
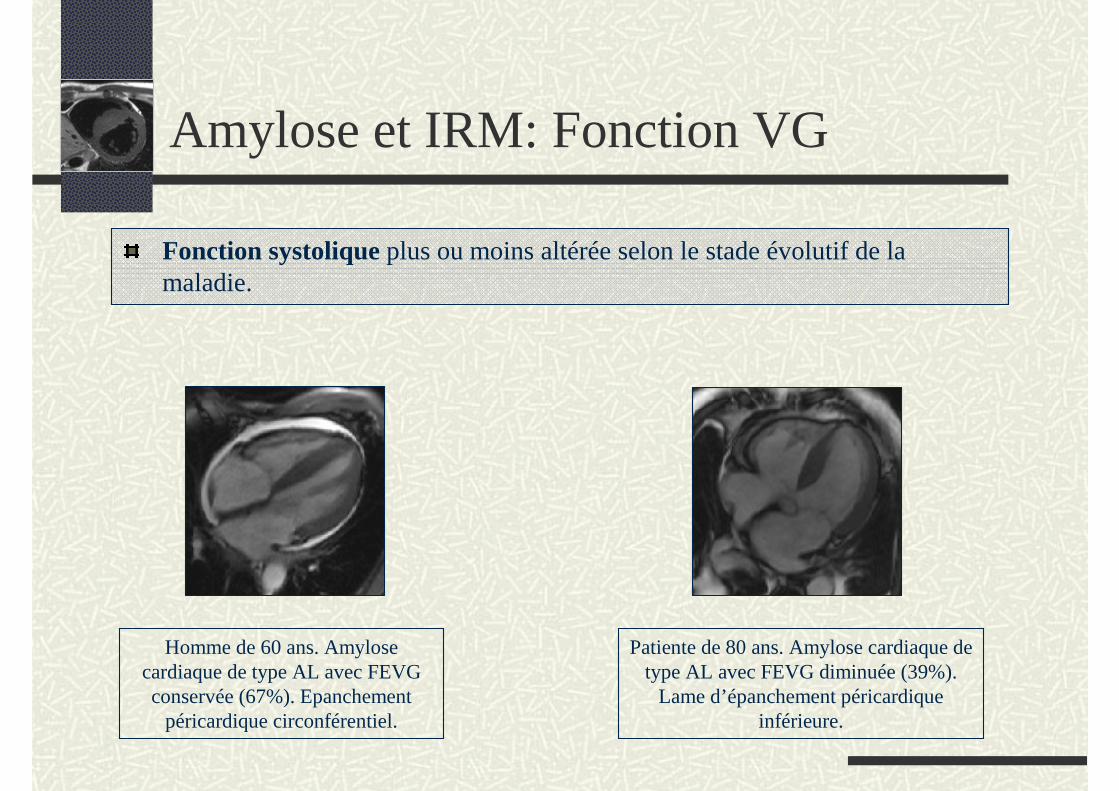
Fonction systolique plus ou moins altérée selon le stade évolutif de la maladie.
Homme de 60 ans. Amylose cardiaque de type AL avec FEVG conservée (67%). Epanchement
péricardique circonférentiel.
Patiente de 80 ans. Amylose cardiaque de type AL avec FEVG diminuée (39%).
Lame d’épanchement péricardique inférieure.
Amylose et IRM: Fonction VG
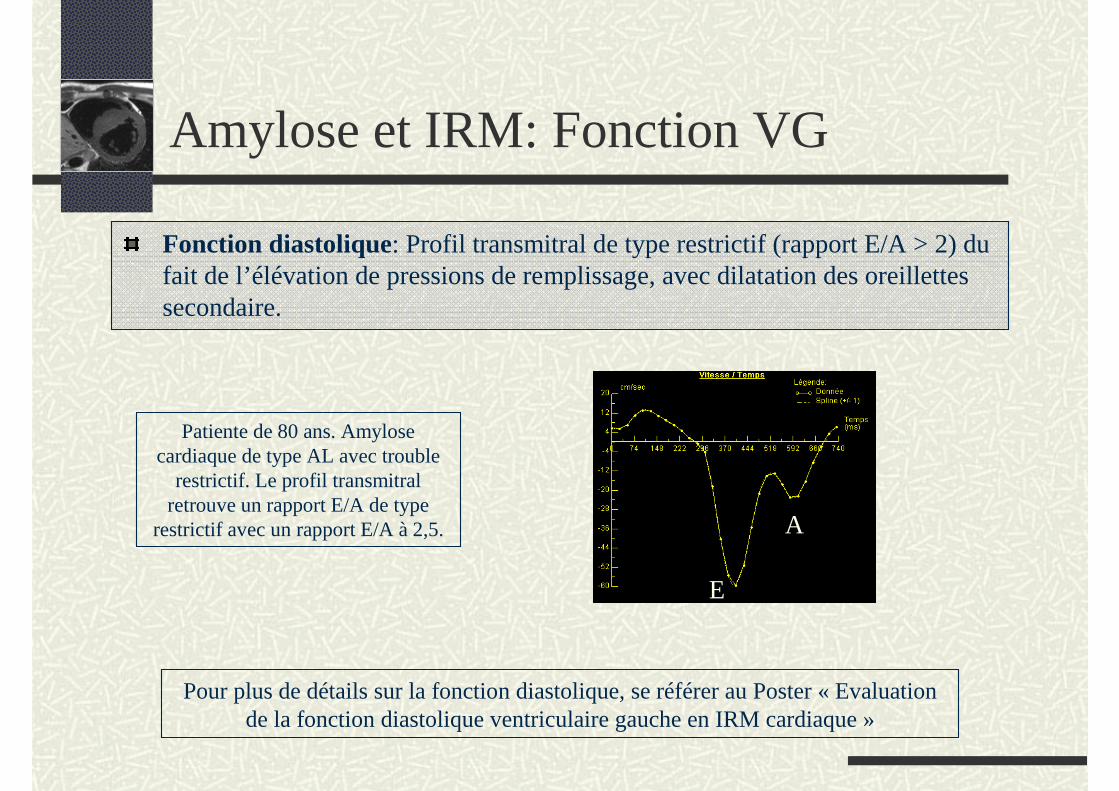
Fonction diastolique: Profil transmitral de type restrictif (rapport E/A > 2) du fait de l’élévation de pressions de remplissage, avec dilatation des oreillettes secondaire.
E
A
Patiente de 80 ans. Amylose cardiaque de type AL avec trouble
restrictif. Le profil transmitral retrouve un rapport E/A de type
restrictif avec un rapport E/A à 2,5.
Pour plus de détails sur la fonction diastolique, se référer au Poster « Evaluation de la fonction diastolique ventriculaire gauche en IRM cardiaque »

Amylose et IRM: Perfusion
Au repos, la perfusion premier passage est souvent normale. Néanmoins, une hypoperfusion sous endocardique peut être présente du fait d’un envahissement fréquent des vaisseaux sous-endocardiques par la protéine amyloïde, conduisant à une diminution de la perfusion sous endocardique (46).
Patiente de 80 ans. Amylose cardiaque de type AL.
Perfusion premier passage retrouvant une hypoperfusion sous
endocardique très marquée.
Amylose et IRM: Rehaussement Tardif
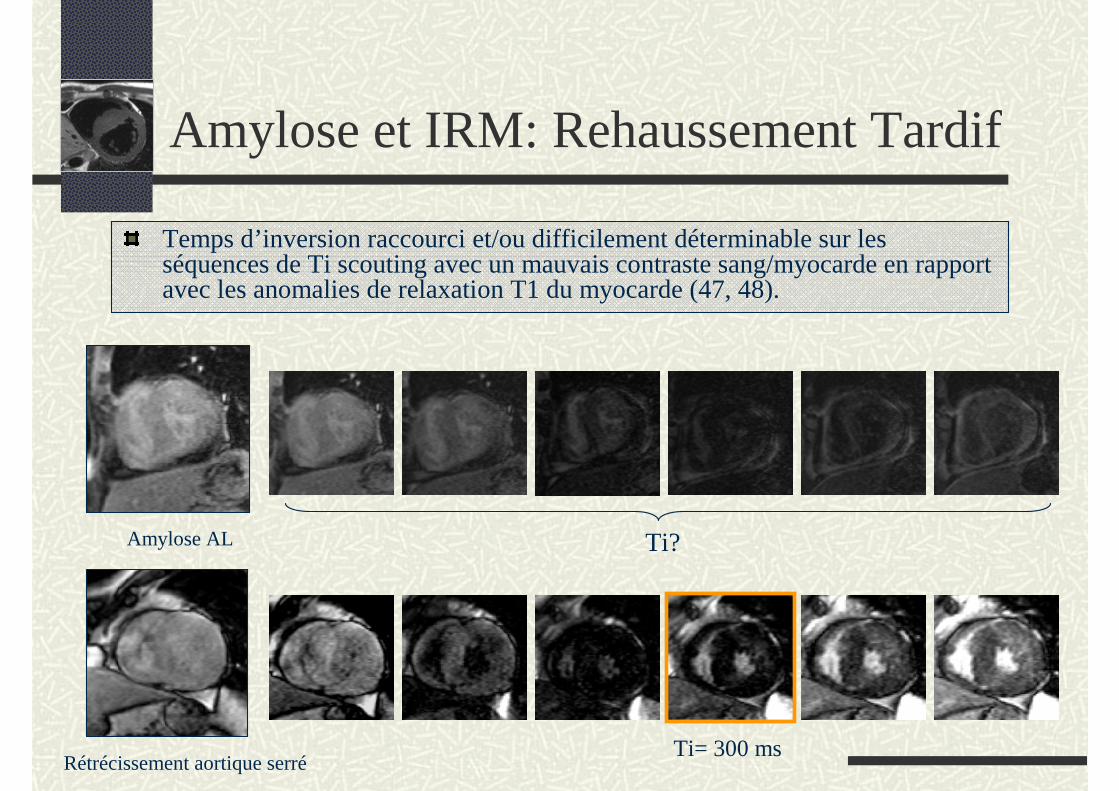
Amylose AL
Temps d’inversion raccourci et/ou difficilement déterminable sur les séquences de Ti scouting avec un mauvais contraste sang/myocarde en rapport avec les anomalies de relaxation T1 du myocarde (47, 48).
Rétrécissement aortique serréTi= 300 ms
Ti?

Amylose et IRM: Rehaussement Tardif
Aspect de rehaussement tardif typique, diffus, atteignant les ventricules et les oreillettes, sans systématisation vasculaire, et de topographie:
plutôt sous endocardique si la séquence est réalisée précocement (5 min) (47, 48).plus hétérogène, diffus et patchy, si elle est réalisée plus tardivement (15 min) (49).
Amylose cardiaque AL – RT à 15 min, diffus, hétérogène intéressant VG, VD, OG et OD.
Amylose cardiaque AL – RT à 5 min, prédominant en sous endocardique

Amylose et IRM: Points Clefs
Au niveau morphologiqueHVG concentrique avec une cavité ventriculaire gauche de petite taille.Epaississement de la paroi libre de l’OD (>6mm) et du septum inter-auriculaire (>6mm).Epaississement de la paroi libre du VD, diffuse (alors que plutôt apicale dans les CMH).Dilatation bi auriculaire.Epanchements péricardiques et pleuraux fréquents.
Au niveau fonctionnelFonction systolique plus ou moins altérée selon le stade évolutif de la maladie.Fonction diastolique: Profil trans-mitral de type restrictif (rapport E/A>2) du fait de l’élévation des pressions de remplissage.
Perfusion premier passageLe plus souvent normale mais possible hypoperfusion sous-endocardique.
Rehaussement TardifTi difficilement déterminable.RT prédominant:
en sous endocardique si la séquence est réalisée précocement (5 min), plus ou moins transmural, avec une atteinte des muscles papillaires. plus hétérogène, diffus et patchy, si elle est réalisée plus tardivement (15 min).

Rétrécissement Aortique: Généralités
Valvulopathie la plus fréquente dans les pays occidentaux (50, 51)
Cause dégénérative calcifiée (maladie de Monckeberg), nettement plus fréquente que le RA sur bicuspidie ou secondaire au rhumatisme articulaire aigu.
ETT +++: examen fondamental permettant d’analyser la fonction VG (systolique et diastolique), d’apprécier l’HVG, de calculer la surface valvulaire aortique grâce à l’équation de continuité, et d’estimer les pressions droites.
Cathétérisme cardiaque: permet l’étude de la fonction VG, de la surface valvulaire aortique par la formule de Gorlin et surtout l’analyse des coronaires en préopératoire.
L’intérêt de l’IRM dans l’exploration des RA a été évalué par de nombreuses études depuis quelques années: l’IRM permet d’une part une étude morphologique et fonctionnelle du VG mais également le calcul de la planimétrie valvulaire aortique. Elle est par contre nettement moins efficace que le scanner pour l’étude des calcifications valvulaires et des artères coronaires.
RA et IRM: Morphologie et Fonction VG
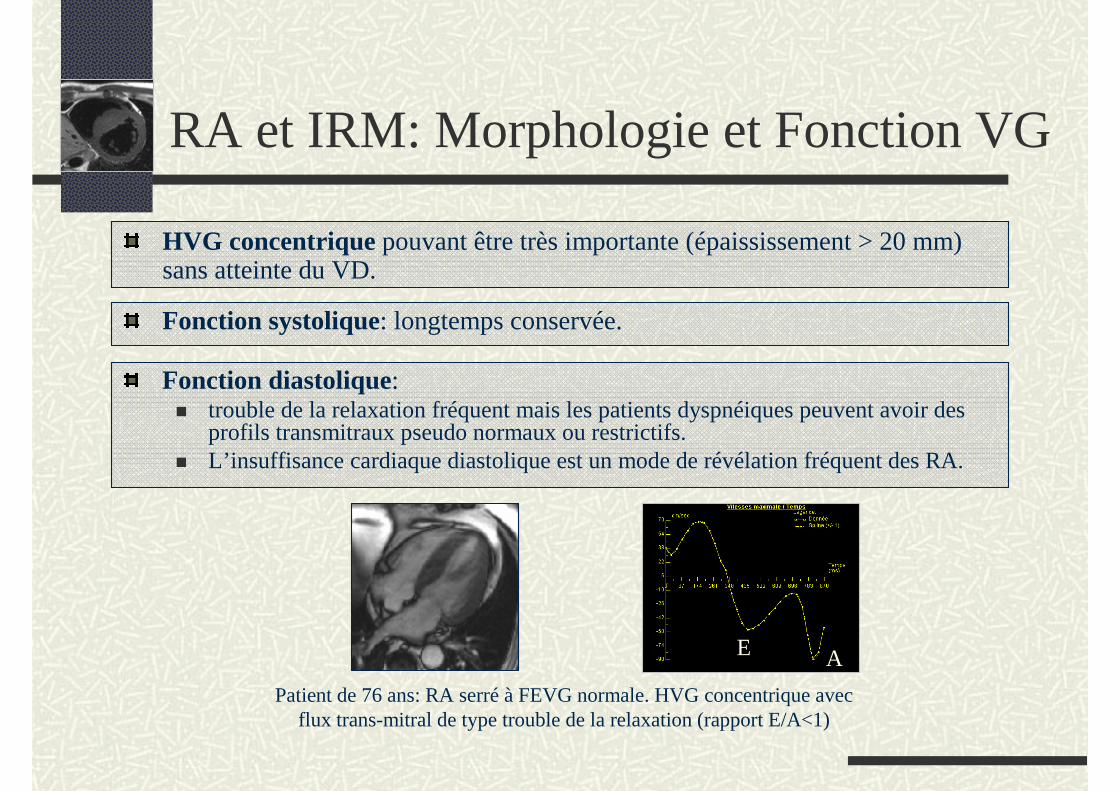
Fonction systolique: longtemps conservée.
Fonction diastolique: trouble de la relaxation fréquent mais les patients dyspnéiques peuvent avoir des profils transmitraux pseudo normaux ou restrictifs. L’insuffisance cardiaque diastolique est un mode de révélation fréquent des RA.
HVG concentrique pouvant être très importante (épaississement > 20 mm) sans atteinte du VD.
Patient de 76 ans: RA serré à FEVG normale. HVG concentrique avec flux trans-mitral de type trouble de la relaxation (rapport E/A<1)
E A
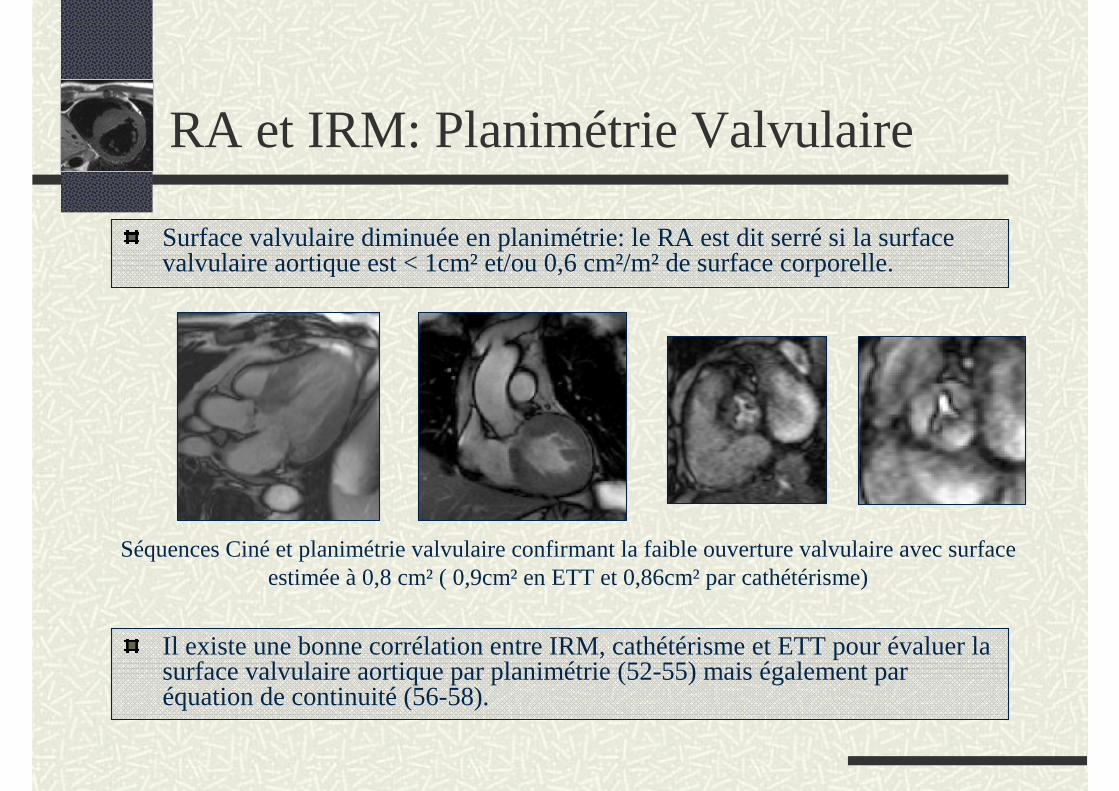
RA et IRM: Planimétrie Valvulaire
Surface valvulaire diminuée en planimétrie: le RA est dit serré si la surface valvulaire aortique est < 1cm² et/ou 0,6 cm²/m² de surface corporelle.
Il existe une bonne corrélation entre IRM, cathétérisme et ETT pour évaluer la surface valvulaire aortique par planimétrie (52-55) mais également par équation de continuité (56-58).
Séquences Ciné et planimétrie valvulaire confirmant la faible ouverture valvulaire avec surface estimée à 0,8 cm² ( 0,9cm² en ETT et 0,86cm² par cathétérisme)

RA et IRM: Perfusion et Rehaussement Tardif
La perfusion premier passage est le plus souvent normale.On peut retrouver dans environ 25% des cas un rehaussement patchy, hétérogène, interstitiel, non systématisé dans un territoire vasculaire, surtout en cas d’épaississement pariétal > 18 mm (59).L’élévation de la post charge entraine en effet une HVG adaptatrice avec dégénérescence myocytaire et fibrose, à l’origine du RT (60, 61).Il faut néanmoins noter que seules les cicatrices fibreuses focalisées peuvent être mises en évidence alors qu’à l’inverse, dans les cas d’infiltrations fibreuses interstitielles diffuses, la technique du RT est prise en défaut (elle est en effet sensible aux différences régionales de concentration en gadolinium).
Patient de 76 ans ayant un rétrécissement aortique serré. RT typique, interstitiel, patchy, non
systématisé.

RA et IRM: Points clefs
Au niveau morphologiqueHVG concentrique pouvant être très importante (> 20 mm).Pas d’atteinte des autres cavités.
Au niveau fonctionnelFonction systolique: longtemps conservée.Fonction diastolique: trouble de la relaxation.Surface valvulaire diminuée en planimétrie et gradients trans-valvulaire augmentés en contraste de phase.
Perfusion premier passageNormale ou trouble de la perfusion sous endocardique
Rehaussement tardifPossible rehaussement patchy hétérogène interstitiel dans les zones les plus hypertrophiées.

HVG du Sportif: Généralités
Les morts subites sont rares chez les sportifs (< 1/200.000).
> 90% des décès surviennent pendant l’entrainement ou la compétition.
Les causes les plus fréquentes de morts subites d’origine cardiovasculaire chez les athlètes sont (62-64):
Les CMH +++
Les anomalies de naissance des coronaires
La DAVD
Autres : myocardites, rétrécissements aortiques congénitaux, ponts intra myocardiques des artères coronaires, syndrome du QT long, CMD…
Néanmoins, si la CMH est la cause la plus fréquente de décès, sa prévalence reste très rare parmi les athlètes (65, 66).
Le problème du dépistage de ces anomalies est donc essentiel et repose sur un screening dont les modalités sont variables d’un pays à l’autre (64, 67).

HVG du Sportif: Dépistage
Quel dépistage réaliser chez le sportif ? (67)
Le screening à l’italienne basé sur les antécédents personnels et familiaux de l’athlète, l’examen clinique et l’ECG 12 dérivations a fait preuve de son efficacité (68, 69).
Ce 1er screening permet d’éliminer une pathologie cardiaque chez la majorité des athlètes, les autres ayant alors un bilan plus exhaustif comportant au minimum une échocardiographie.
Parmi les patients qui vont bénéficier d’un 2ème screening, le plus souvent du fait d’anomalies ECG, très peu auront au final une maladie cardiaque, en particulier une CMH (66).
Aux USA, le 1er screening ne comporte pas d’ECG pour des raisons économiques (64).

HVG du Sportif: Dépistage
En pratique, la difficulté de ce dépistage vient du fait que l’adaptation du cœur à l’effort peut mimer des pathologies telles que la CMH et la CMD car 2% des athlètes très entrainés ont (70):
Une HVG (avec une épaisseur ≥ 13 mm mais habituellement < 16 mm). Si ≥ 16 mm on parle de CMH, même chez un sportif, et un bilan complémentaire doit être effectué (7).
Une augmentation de la masse VG.
Une augmentation des volumes du VG et de l’OG.
Ces constatations peuvent toucher également le VD.

HVG du Sportif: Dépistage
Quels arguments permettent, lors de ce dépistage, d’affirmer le caractère physiologique d’une HVG?
Pas d’antécédent familial au 1er degré de CMH.
Pas d’anomalie à l’examen clinique, en particulier pas de souffle cardiaque.
ECG normal (71-75) en sachant que environ 25% des athlètes ont des anomalies ECG sans signification pathologique, liées à l’adaptation du cœur à l’effort (76).
Pas d’obstruction intra ventriculaire avec mouvement systolique antérieur de la valve mitrale (SAM) (77).
Fonction diastolique normale en particulier pas de trouble de la relaxation au niveau du flux transmitral dans une HVG physiologique (78).
Augmentation du volume du VG proportionnel à l’augmentation de l’épaisseur pariétale car l’HVG normale de l’athlète s’associe à une dilatation de la cavité VG (79).
Test d’effort métabolique normal (80).
1er
screening
2ème
screening
Dans les cas douteux, l’arrêt du sport permet pour la grande majorité des athlètes une baisse de l’HVG et des volumes ventriculaires, confirmant l’HVG physiologique (81-86).

HVG du Sportif et IRM
Quelle place pour l’IRM?
La place de l’IRM dans ce dépistage reste à définir même si l’on sait qu’elle est utile pour éliminer une CMH apicale, situation dans laquelle l’ETT est nettement prise à défaut (85).
Son rôle en 3ème intention, en complément de l’ETT, pour rechercher des arguments en faveur d’une CMH, semble attractif car elle permet:
La quantification exhaustive de l’épaisseur pariétale et de la masse VG.
La quantification précise des volumes myocardiques.
L’étude de la perfusion myocardique et la recherche de rehaussement tardif.
L’autre avantage évident de l’IRM est son potentiel pour dépister les autres causes de mort subite telles que la DAVD, les myocardites, les anomalies de naissance des coronaires, les CMD et les RA congénitaux.
HVG du Sportif et IRM
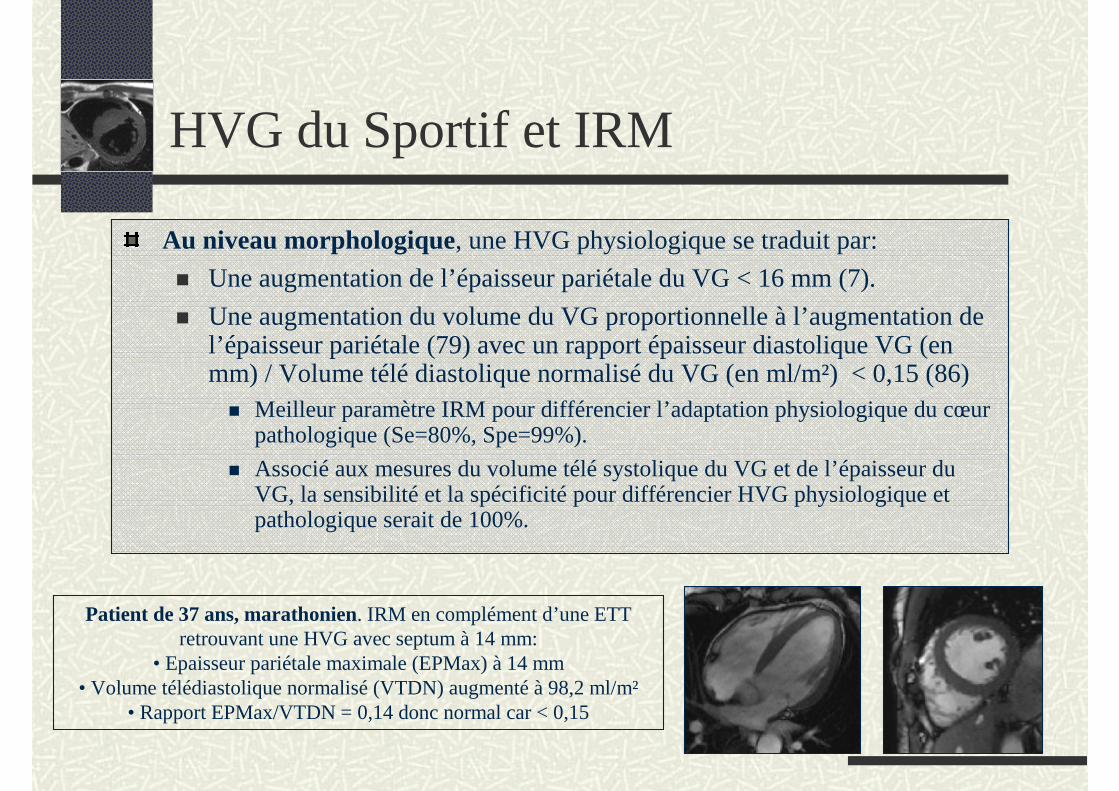
Au niveau morphologique, une HVG physiologique se traduit par:
Une augmentation de l’épaisseur pariétale du VG < 16 mm (7).
Une augmentation du volume du VG proportionnelle à l’augmentation de l’épaisseur pariétale (79) avec un rapport épaisseur diastolique VG (en mm) / Volume télé diastolique normalisé du VG (en ml/m²) < 0,15 (86)
Meilleur paramètre IRM pour différencier l’adaptation physiologique du cœur pathologique (Se=80%, Spe=99%).
Associé aux mesures du volume télé systolique du VG et de l’épaisseur du VG, la sensibilité et la spécificité pour différencier HVG physiologique et pathologique serait de 100%.
Patient de 37 ans, marathonien. IRM en complément d’une ETT retrouvant une HVG avec septum à 14 mm:
• Epaisseur pariétale maximale (EPMax) à 14 mm• Volume télédiastolique normalisé (VTDN) augmenté à 98,2 ml/m²
• Rapport EPMax/VTDN = 0,14 donc normal car < 0,15
HVG du Sportif et IRM
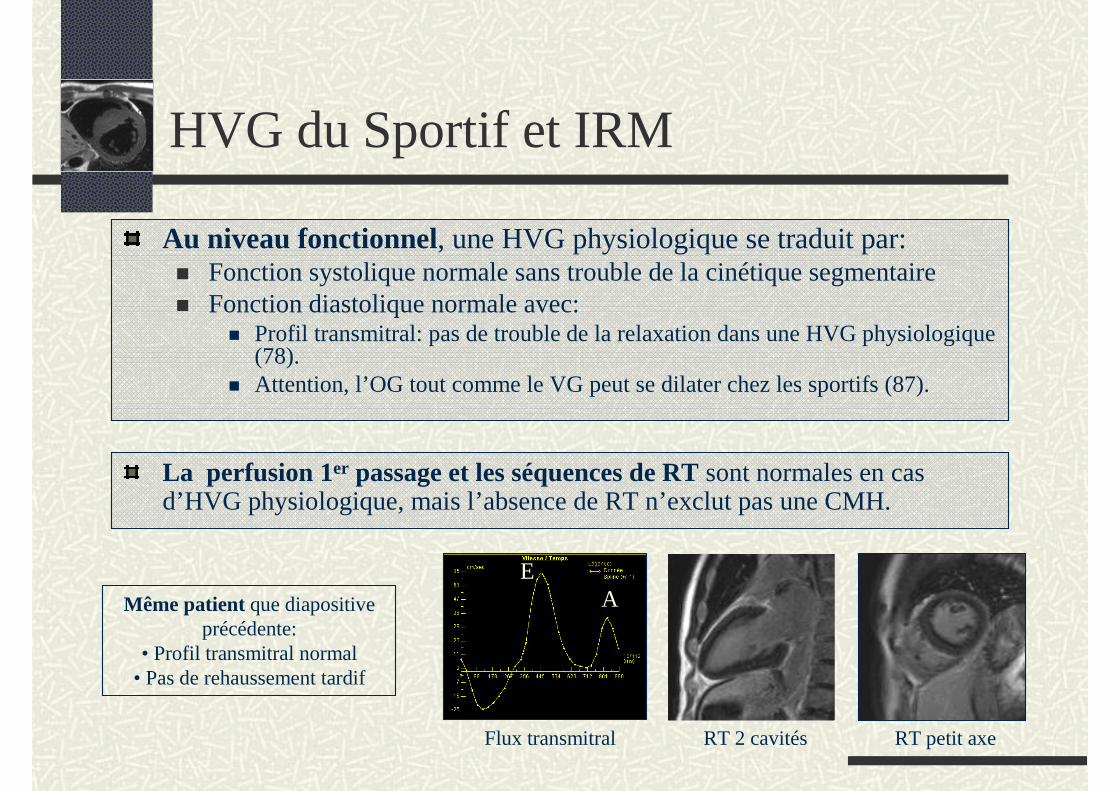
Au niveau fonctionnel, une HVG physiologique se traduit par:Fonction systolique normale sans trouble de la cinétique segmentaireFonction diastolique normale avec:
Profil transmitral: pas de trouble de la relaxation dans une HVG physiologique (78).Attention, l’OG tout comme le VG peut se dilater chez les sportifs (87).
La perfusion 1er passage et les séquences de RT sont normales en cas d’HVG physiologique, mais l’absence de RT n’exclut pas une CMH.
Même patient que diapositive précédente:
• Profil transmitral normal• Pas de rehaussement tardif
Flux transmitral RT 2 cavités RT petit axe
EA

HVG du Sportif et IRM: Points Clefs
Au niveau morphologiqueHVG modérée avec épaisseur pariétale maximale < 16 mm.Augmentation du volume télédiastolique VG proportionnelle à l’augmentation de l’épaisseur pariétale du VG.Rapport épaisseur diastolique VG (en mm) / Volume télé diastolique normalisé du VG (en ml/m²) < 0,15.
Au niveau fonctionnelFEVG normale sans trouble de la cinétique segmentaire.Pas de dysfonction diastolique, en particulier pas de trouble de la relaxation au flux trans mitral. Se méfier du volume de l’OG qui peut être augmenté tout comme le volume du VG.
Perfusion premier passageNormale.
Rehaussement tardifAbsent mais ce critère n’élimine pas une CMH

Sarcoïdose Cardiaque: Généralités
Granulomatose systémique avec atteinte cardiaque intéressant environ 25% des patients dans les séries autopsiques (88) alors que seulement 5% des patients présentent des signes cliniques et ECG de localisation myocardique de leur vivant (89).
L’atteinte cardiaque est pourtant responsable de la majorité des décès en rapport avec la sarcoïdose (troubles du rythme ou de la conduction pouvant entrainer des morts subites en particulier).
Il faut donc identifier l’atteinte cardiaque avant qu’elle ne soit symptomatique, pour initier un traitement corticoïde qui améliore la fonction VG et prévient les troubles du rythme.
La sensibilité de l’ETT, de l’ECG et de la scintigraphie est nettement insuffisante pour infirmer une atteinte cardiaque. De même, les biopsies endomyocardiques montrant les granulomes non caséeux manquent de sensibilité et sont invasives donc rarement réalisées en pratique (90).
Sarcoïdose Cardiaque et IRM
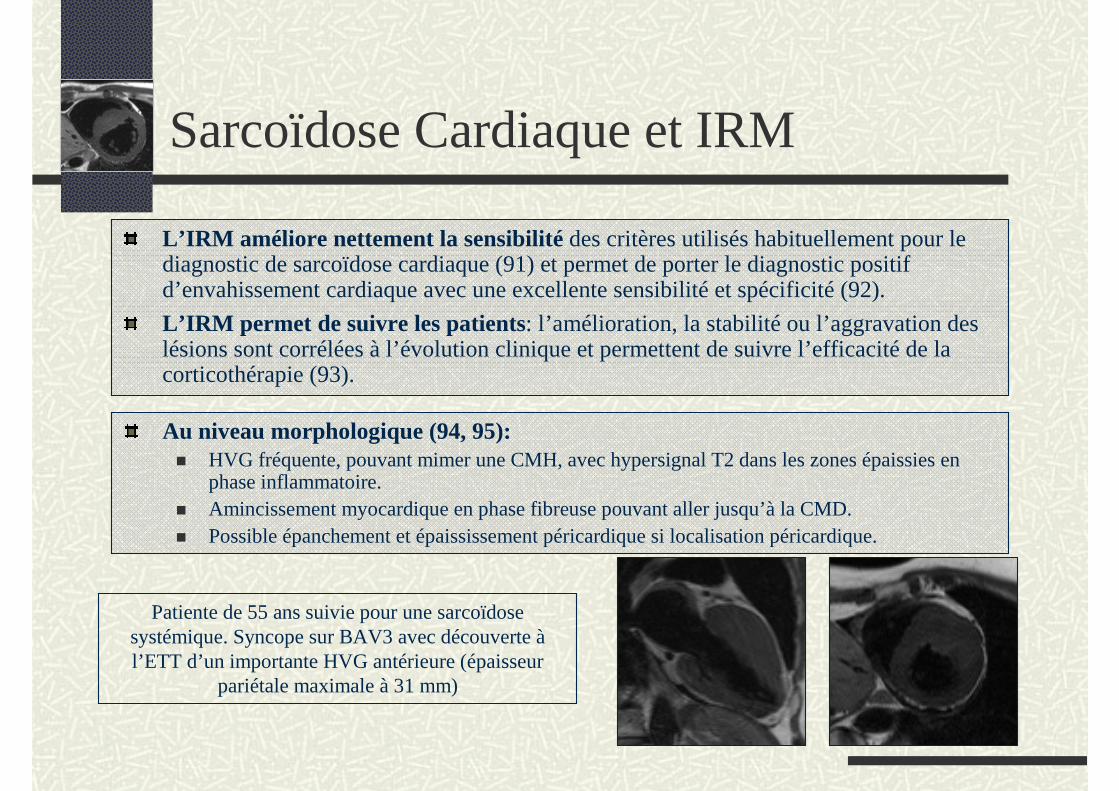
L’IRM améliore nettement la sensibilité des critères utilisés habituellement pour le diagnostic de sarcoïdose cardiaque (91) et permet de porter le diagnostic positif d’envahissement cardiaque avec une excellente sensibilité et spécificité (92).
L’IRM permet de suivre les patients: l’amélioration, la stabilité ou l’aggravation des lésions sont corrélées à l’évolution clinique et permettent de suivre l’efficacité de la corticothérapie (93).
Patiente de 55 ans suivie pour une sarcoïdose systémique. Syncope sur BAV3 avec découverte àl’ETT d’un importante HVG antérieure (épaisseur
pariétale maximale à 31 mm)
Au niveau morphologique (94, 95):HVG fréquente, pouvant mimer une CMH, avec hypersignal T2 dans les zones épaissies en phase inflammatoire.Amincissement myocardique en phase fibreuse pouvant aller jusqu’à la CMD.Possible épanchement et épaississement péricardique si localisation péricardique.
Sarcoïdose Cardiaque et IRM
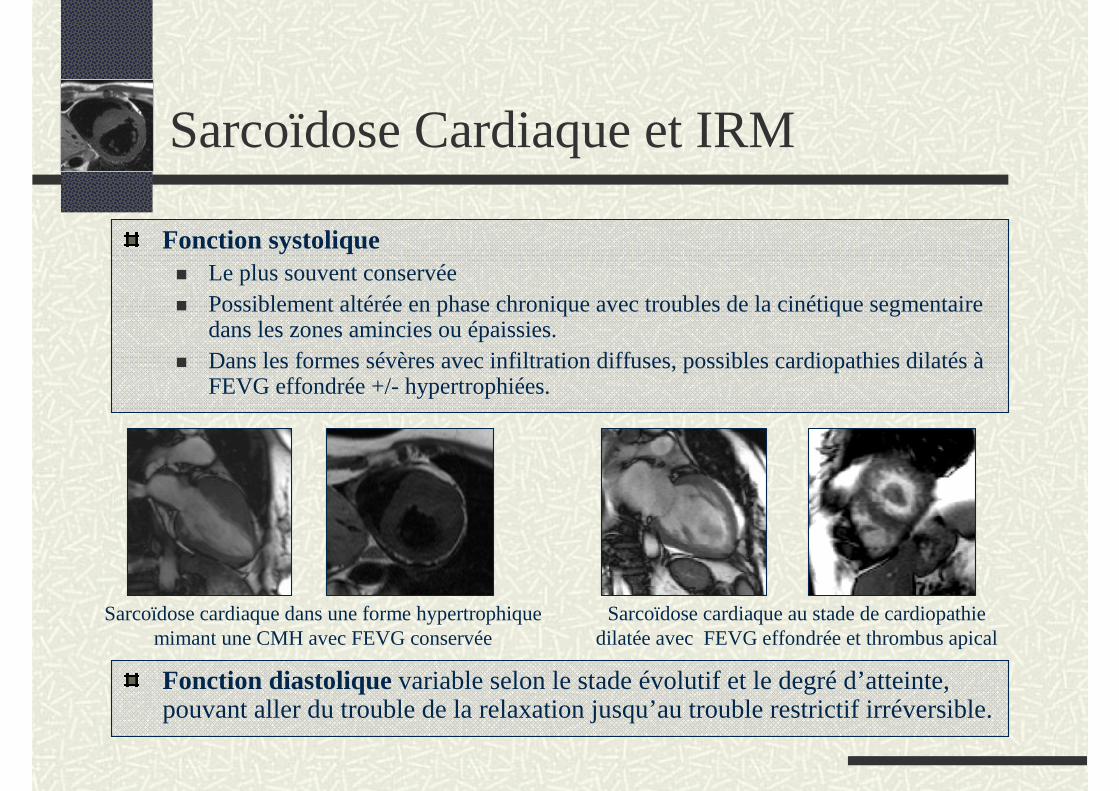
Fonction systoliqueLe plus souvent conservéePossiblement altérée en phase chronique avec troubles de la cinétique segmentaire dans les zones amincies ou épaissies. Dans les formes sévères avec infiltration diffuses, possibles cardiopathies dilatés àFEVG effondrée +/- hypertrophiées.
Fonction diastolique variable selon le stade évolutif et le degré d’atteinte, pouvant aller du trouble de la relaxation jusqu’au trouble restrictif irréversible.
Sarcoïdose cardiaque dans une forme hypertrophique mimant une CMH avec FEVG conservée
Sarcoïdose cardiaque au stade de cardiopathie dilatée avec FEVG effondrée et thrombus apical
Sarcoïdose Cardiaque et IRM
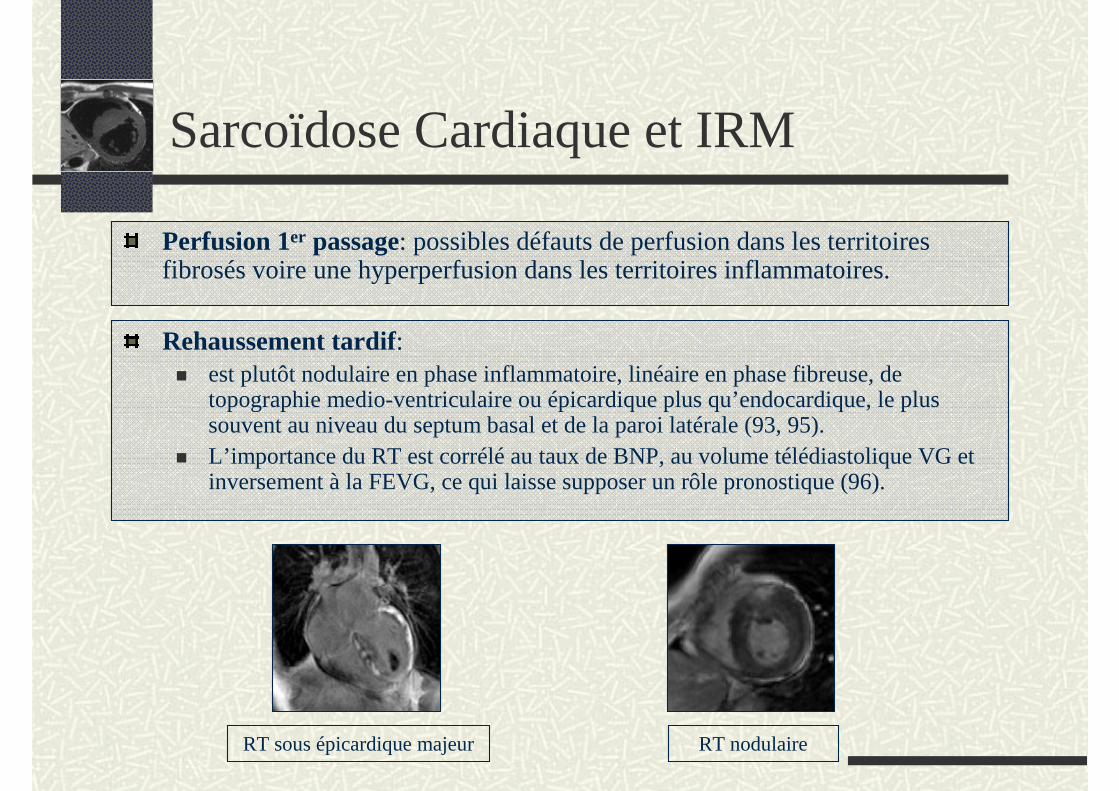
Rehaussement tardif:est plutôt nodulaire en phase inflammatoire, linéaire en phase fibreuse, de topographie medio-ventriculaire ou épicardique plus qu’endocardique, le plus souvent au niveau du septum basal et de la paroi latérale (93, 95).L’importance du RT est corrélé au taux de BNP, au volume télédiastolique VG et inversement à la FEVG, ce qui laisse supposer un rôle pronostique (96).
Perfusion 1er passage: possibles défauts de perfusion dans les territoires fibrosés voire une hyperperfusion dans les territoires inflammatoires.
RT sous épicardique majeur RT nodulaire
Sarcoïdose Cardiaque et IRM
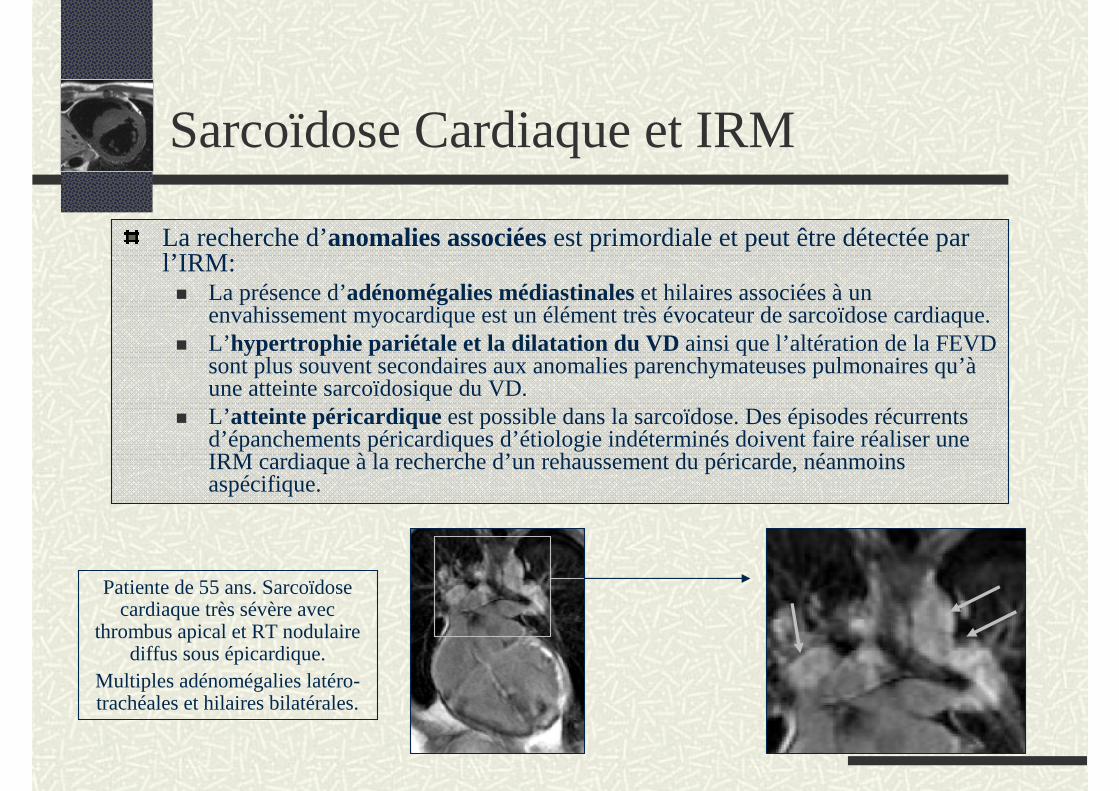
La recherche d’anomalies associées est primordiale et peut être détectée par l’IRM:
La présence d’adénomégalies médiastinales et hilaires associées à un envahissement myocardique est un élément très évocateur de sarcoïdose cardiaque.L’hypertrophie pariétale et la dilatation du VD ainsi que l’altération de la FEVD sont plus souvent secondaires aux anomalies parenchymateuses pulmonaires qu’àune atteinte sarcoïdosique du VD. L’atteinte péricardique est possible dans la sarcoïdose. Des épisodes récurrents d’épanchements péricardiques d’étiologie indéterminés doivent faire réaliser une IRM cardiaque à la recherche d’un rehaussement du péricarde, néanmoins aspécifique.
Patiente de 55 ans. Sarcoïdose cardiaque très sévère avec
thrombus apical et RT nodulaire diffus sous épicardique.
Multiples adénomégalies latéro-trachéales et hilaires bilatérales.

Sarcoïdose Cardiaque et IRM
Quand réaliser une IRM chez un patient sarcoïdosique?Les « recommandations » actuelles, non émises par les sociétés savantes (97), ne tiennent pas compte de l’IRM pour le diagnostic de sarcoïdose cardiaque. Elles manquent par ailleurs de sensibilité et nécessitent la présence d’anomalies ECG dont on sait qu’elles traduisent une atteinte myocardique déjà sévère.L’étude publiée par l’équipe de la Pitié Salpêtrière (98) est en ce sens un premier pas vers la clarification du rôle de l’IRM dans l’atteinte cardiaque de la sarcoïdose et peut être résumée ainsi:
En cas de sarcoïdose peu sévère (articulaire, cutanée, syndrome de Löfgren, atteinte des glandes exocrines): on peut se limiter à l’examen clinique et àl’ECG.Au cours de sarcoïdose très active (signes généraux sévères, atteinte pulmonaire évolutive, diffusion multisystémique du processus granulomateux), le bilan cardiologique est légitime. Ce dernier devient indispensable en présence de localisations rares (neurologique, osseuse, ORL, rénale…).
Lorsqu’un bilan cardiologique est décidé, il doit être exhaustif: ETT, scintigraphie, IRM cardiaque.

Sarcoïdose Cardiaque et IRM: Points Clefs
Au niveau morphologique
Epaississement myocardique en phase inflammatoire (pouvant mimer une CMH) avec hypersignal T2 dans les zones épaissies.
Amincissement myocardique en phase fibreuse pouvant aller jusqu’à la CMD.
Possible épanchement et épaississement péricardique si localisation péricardique.
Au niveau fonctionnel
Fonction systolique le plus souvent conservée mais possiblement altérée en phase chronique avec troubles de la cinétique segmentaire dans les zones amincies ou épaissies.
Fonction diastolique variable selon le stade évolutif et le degré d’atteinte.
Perfusion premier passage
possibles défauts de perfusion dans les territoires fibrosés, hyperperfusion dans les territoires inflammatoires.
Rehaussement tardif
Plutôt nodulaire en phase inflammatoire, linéaire en phase fibreuse, de topographie medio-ventriculaire ou épicardique plus qu’endocardique, le plus souvent au niveau du septum basal et de la paroi latérale.

Maladie de Fabry: Généralités
Pathologie héréditaire du métabolisme des glycosphingolipides due à un déficit en enzyme lysosomale (α-galactosidase A), avec accumulation du substrat non dégradé dans les tissus (99, 100).
Transmission récessive liée au chromosome X donc atteinte surtout des hommes. Les femmes hétérozygotes ont une vie quasiment normale avec quelques manifestations au fur et à mesure de leur vieillissement, rarement des complications aussi graves que dans la forme classique des hommes.
Chez les hommes, la façon la plus fiable de faire le diagnostic est la mesure de l'activité enzymatique de l'alpha-galactosidase dans les leucocytes.
La détection de la mutation est possible chez près de 100% des hommes malades et plus de 250 mutations ont été décrites.
Traitement enzymatique substitutif efficace (101-103) avec diminution significative de la masse cardiaque évaluée par IRM (104).

Maladie de Fabry: Généralités
Maladie rare bien que probablement sous évaluée (< 1 naissance / 50 000 garçons).
L’évaluation de la prévalence chez les patients suivis pour CMH est variable selon les études et la population étudiée (105, 106) mais une étude récente sur 508 patients (107) retrouvait une prévalence d’environ 1% dans une population ciblée de patients suivis pour CMH. Les conséquences de la découverte d’une maladie de Fabry sont importantes pour le traitement et le dépistage familial.
Manifestations systémiques variées, les plus graves étant rénales (insuffisance rénale), neurologiques (AVC) et myocardiques (99, 100).
Au niveau cardiaque, les manifestations sont variées: HVG +++, raccourcissement de l’intervalle PR mais aussi épaississements valvulaires aortique et mitral avec possible IM par prolapsus, douleurs angineuses par atteinte microvasculaire, BAV et troubles du rythme avec morts subites (100, 108).
Maladie de Fabry et IRM
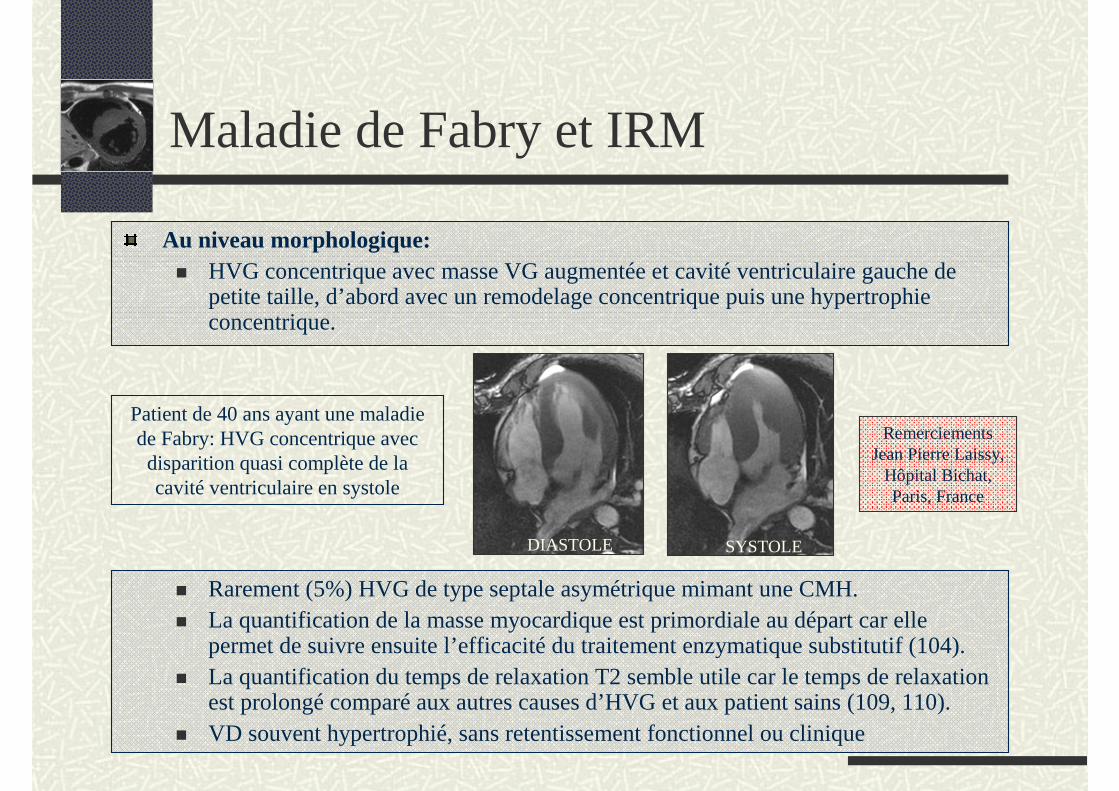
Au niveau morphologique:HVG concentrique avec masse VG augmentée et cavité ventriculaire gauche de petite taille, d’abord avec un remodelage concentrique puis une hypertrophie concentrique.
Patient de 40 ans ayant une maladie de Fabry: HVG concentrique avec
disparition quasi complète de la cavité ventriculaire en systole
DIASTOLE SYSTOLE
Remerciements Jean Pierre Laissy,
Hôpital Bichat, Paris, France
Rarement (5%) HVG de type septale asymétrique mimant une CMH.La quantification de la masse myocardique est primordiale au départ car elle permet de suivre ensuite l’efficacité du traitement enzymatique substitutif (104).La quantification du temps de relaxation T2 semble utile car le temps de relaxation est prolongé comparé aux autres causes d’HVG et aux patient sains (109, 110).VD souvent hypertrophié, sans retentissement fonctionnel ou clinique

Maladie de Fabry et IRM
Au niveau fonctionnel:Fonction systolique le plus souvent normale.Fonction diastolique altérée, le plus souvent avec trouble de la relaxation mais atteinte restrictive possible en cas de stade évolué avec fibrose importante.
Perfusion 1er passageLe plus souvent normale mais possible réduction de la réserve coronaire par atteinte microvasculaire, retrouvée à l’IRM de stress, avec hypoperfusion des territoires atteints (démontré en PET, pas encore en IRM).
Rehaussement TardifZones de fibrose avec RT le plus souvent linéaires chez l’homme et patchy nodulaire chez la femme, avec une topographie qui n’est pas sous endocardique, contrairement aux infarctus, et une atteinte plus fréquente au niveau inféro-latéro basal (111, 112).

Maladie de Fabry: Points clefs
Au niveau morphologiqueHVG concentrique avec masse VG augmentée et cavité VG de petite taille, d’abord avec un remodelage concentrique puis une hypertrophie concentrique.Temps de relaxation T2 augmenté.VD souvent hypertrophié, sans retentissement fonctionnel ou clinique.
Au niveau fonctionnelFonction systolique le plus souvent normale.Fonction diastolique altérée, le plus souvent avec trouble de la relaxation mais atteinte restrictive possible en cas de stade évolué avec fibrose importante.
Perfusion 1er passageLe plus souvent normale mais possible réduction de la réserve coronaire par atteinte microvasculaire
Rehaussement TardifRT le plus souvent linéaire chez l’homme et patchy nodulaire chez la femme, avec une topographie qui n’est pas sous endocardique, contrairement aux infarctus et une atteinte plus fréquente au niveau inféro-latéro basal

HTA: Généralités
L’HTA est une des causes principales d’HVG.
L’HVG secondaire à l’HTA est symétrique, avec un remodelage concentrique.
Chez les patients hypertendus, l’HVG secondaire est facteur indépendant de morbidité et de mortalité, prédisposant à l’insuffisance cardiaque, aux troubles du rythme ventriculaire, aux AVC ischémiques, à la FA.
A l’hypertrophie myocytaire secondaire à l’élévation de la post charge s’ajoute une fibrose interstitielle et des anomalies de la microcirculation coronaire

HTA et IRM
Au niveau morphologique:HVG concentrique, avec une épaisseur myocardique variable mais symétrique.Masse augmentée chez 28% des patients hypertendus de race blanche et 62% des patients de race noire (113). Il faut noter que l’IRM est nettement plus sensible que l’ETT pour dépister une HVG chez les patients hypertendus (Se ETT=30%) qui elle-même est nettement plus sensible que l’ECG (Se=5 à 10%).
Au niveau fonctionnel:La fonction systolique est le plus souvent normale mais peut être altérée en cas de cardiopathie ischémique associée (l’HTA étant un important facteur de risque cardiovasculaire). Le lien direct entre dysfonction systolique et HTA reste débattu (114-116). Fonction diastolique altérée +++: l’HTA demeure une des causes les plus fréquentes d’insuffisance cardiaque diastolique.

HTA et IRM
Perfusion:Au repos, la perfusion est le plus souvent normale.Sous stress, l’atteinte de la microcirculation coronaire entraine une réduction de la réserve coronaire lors des épreuves pharmacologiques vasodilatatrices, même en l’absence de coronaropathie sous jacente, avec ischémie sous-endocardique (114).La combinaison de ces anomalies de perfusion et de relaxation est à l’origine de la dysfonction diastolique.
Rehaussement tardif:Les séquences de RT sont souvent normales.Néanmoins, du fait de la fibrose interstitielle et de l’attente de la microcirculation, on peut avoir un RT de type interstitiel intramyocardique (117). Comme pour les RA, seules les zones de fibrose focalisées peuvent être mises en évidence.

HTA: Points Clefs
Au niveau morphologiqueHVG concentrique, avec une épaisseur myocardique variable mais symétrique.
Au niveau fonctionnelFonction systolique le plus souvent normale en l’absence de cardiopathie ischémique associée.Fonction diastolique perturbée à des degrés variables, du trouble de la relaxation au trouble de la compliance avec insuffisance cardiaque diastolique.
Perfusion 1er passageNormale au repos, mais possible hypoperfusion sous endocardique par atteinte de la microcirculation pouvant être démasquée sous stress.
Rehaussement TardifLe plus souvent normales mais possible RT interstitiel en cas de fibrose focalisée.
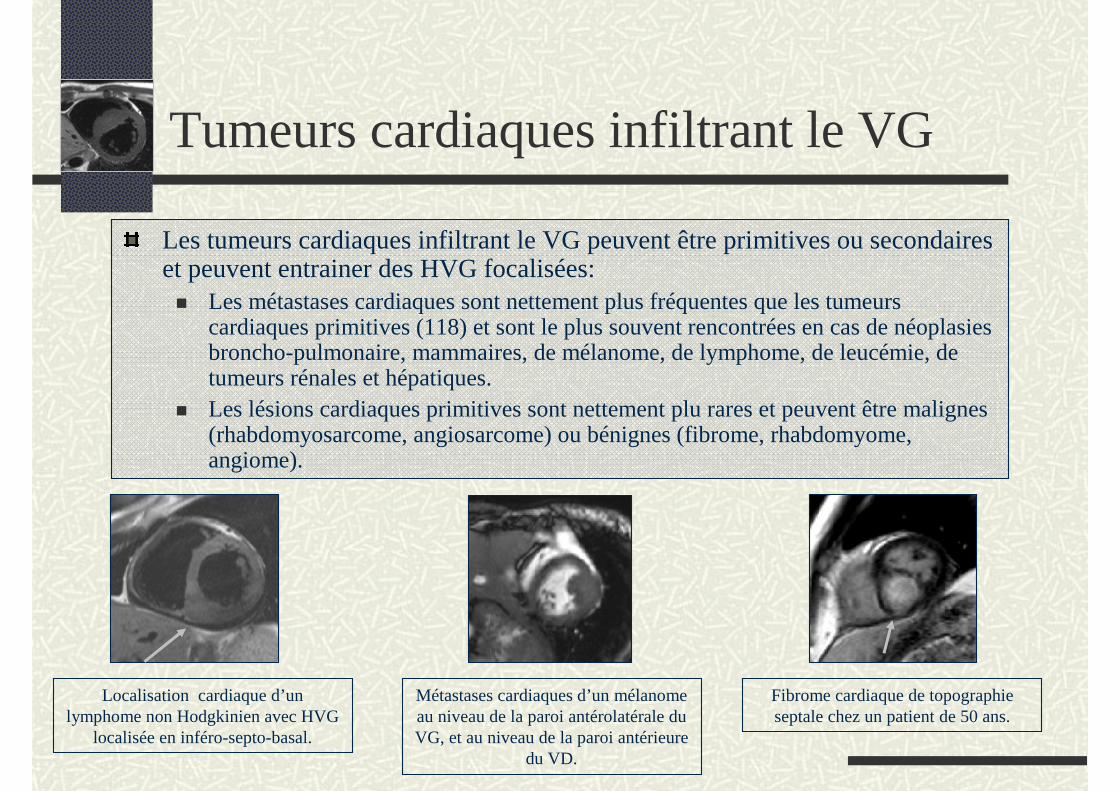
Tumeurs cardiaques infiltrant le VG
Les tumeurs cardiaques infiltrant le VG peuvent être primitives ou secondaires et peuvent entrainer des HVG focalisées:
Les métastases cardiaques sont nettement plus fréquentes que les tumeurs cardiaques primitives (118) et sont le plus souvent rencontrées en cas de néoplasies broncho-pulmonaire, mammaires, de mélanome, de lymphome, de leucémie, de tumeurs rénales et hépatiques.Les lésions cardiaques primitives sont nettement plu rares et peuvent être malignes (rhabdomyosarcome, angiosarcome) ou bénignes (fibrome, rhabdomyome, angiome).
Localisation cardiaque d’un lymphome non Hodgkinien avec HVG
localisée en inféro-septo-basal.
Métastases cardiaques d’un mélanome au niveau de la paroi antérolatérale du VG, et au niveau de la paroi antérieure
du VD.
Fibrome cardiaque de topographie septale chez un patient de 50 ans.
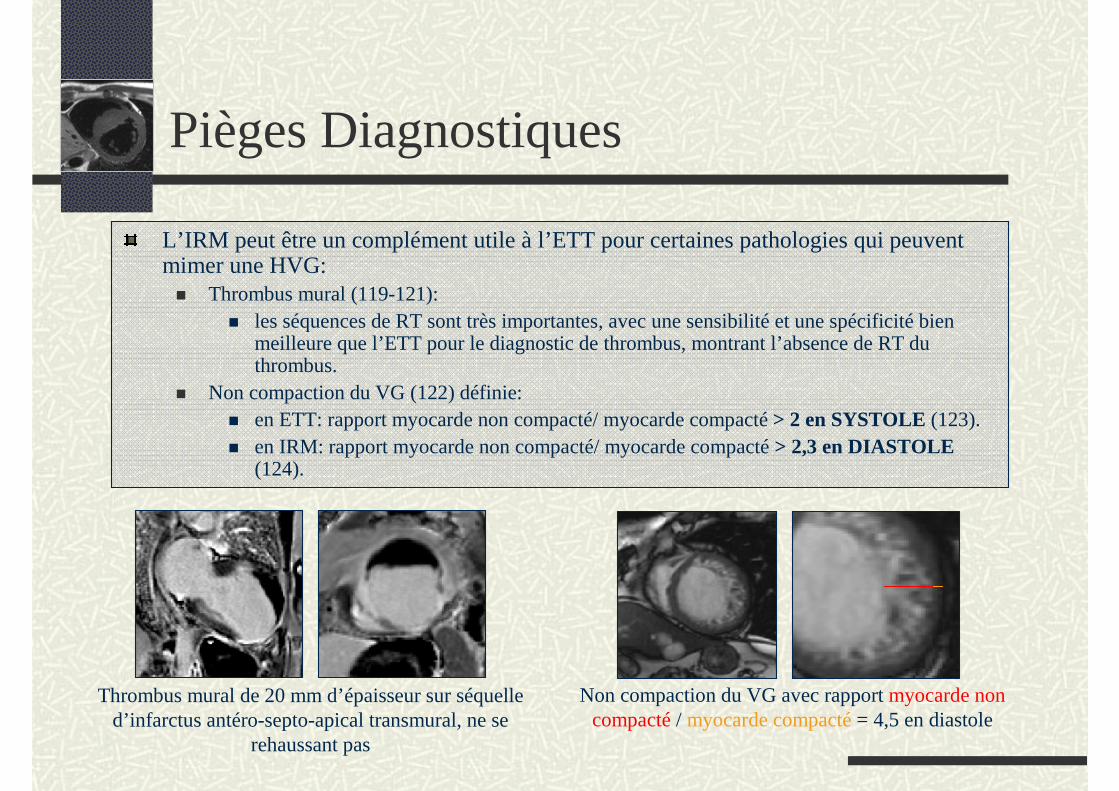
Pièges Diagnostiques
L’IRM peut être un complément utile à l’ETT pour certaines pathologies qui peuvent mimer une HVG:
Thrombus mural (119-121): les séquences de RT sont très importantes, avec une sensibilité et une spécificité bien meilleure que l’ETT pour le diagnostic de thrombus, montrant l’absence de RT du thrombus.
Non compaction du VG (122) définie:en ETT: rapport myocarde non compacté/ myocarde compacté > 2 en SYSTOLE (123).en IRM: rapport myocarde non compacté/ myocarde compacté > 2,3 en DIASTOLE (124).
Thrombus mural de 20 mm d’épaisseur sur séquelle d’infarctus antéro-septo-apical transmural, ne se
rehaussant pas
Non compaction du VG avec rapport myocarde non compacté / myocarde compacté = 4,5 en diastole

Conclusion
L’IRM est utile dans l’exploration des HVG en complément de l’ETT dans certaines indications:
Exploration des CMH.
Suspicion d’amylose cardiaque.
Suspicion de maladie de Fabry.
Exploration des HVG du sportif.
Recherche de localisation cardiaque de la sarcoïdose.
Tumeur cardiaque localisée au VG.
L’IRM est un examen de seconde intention qui permet une étude exhaustive de la morphologie, de la fonction, de la perfusion et du rehaussement du VG.
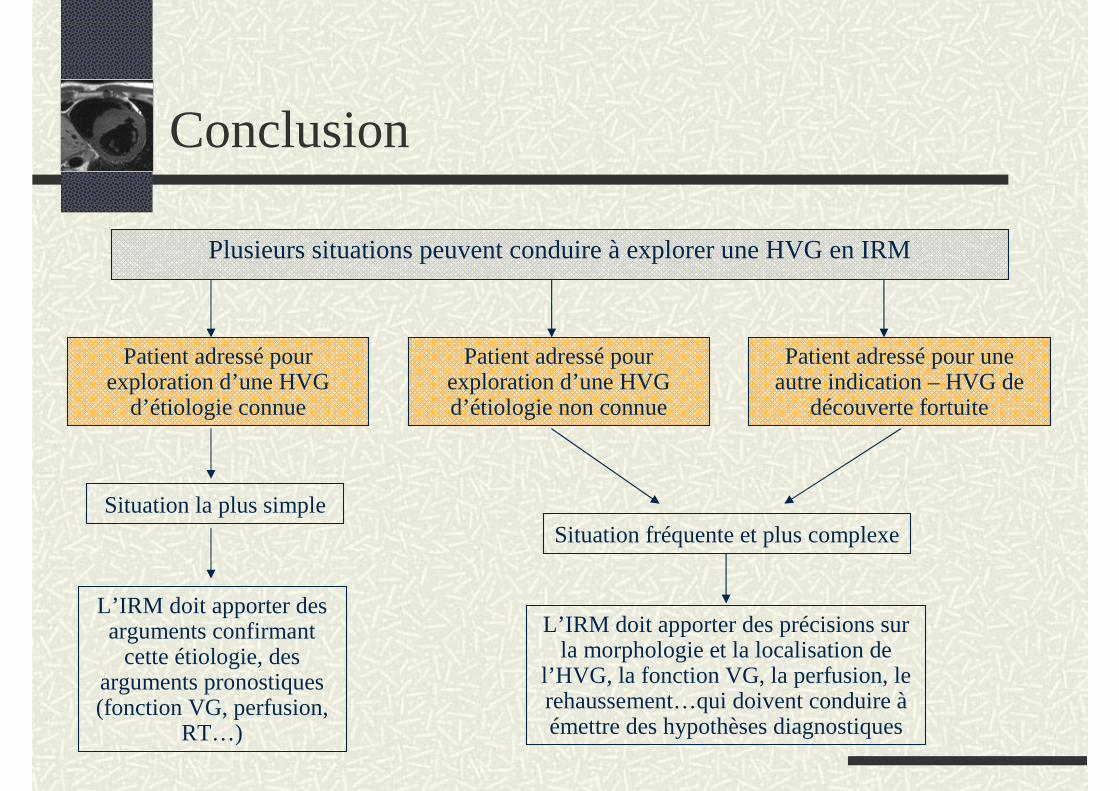
Conclusion
Plusieurs situations peuvent conduire à explorer une HVG en IRM
Patient adressé pour exploration d’une HVG d’étiologie non connue
Patient adressé pour exploration d’une HVG
d’étiologie connue
Patient adressé pour une autre indication – HVG de
découverte fortuite
L’IRM doit apporter des précisions sur la morphologie et la localisation de
l’HVG, la fonction VG, la perfusion, le rehaussement…qui doivent conduire àémettre des hypothèses diagnostiques
L’IRM doit apporter des arguments confirmant
cette étiologie, des arguments pronostiques (fonction VG, perfusion,
RT…)
Situation la plus simpleSituation fréquente et plus complexe
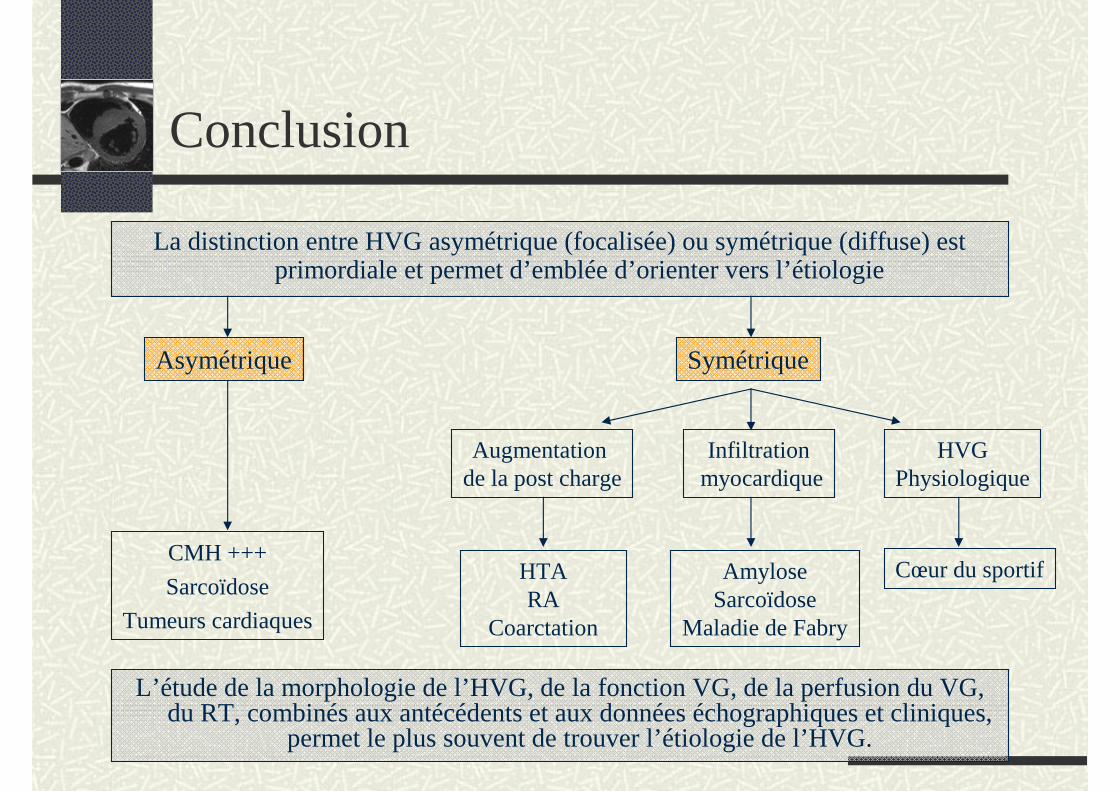
Conclusion
La distinction entre HVG asymétrique (focalisée) ou symétrique (diffuse) est primordiale et permet d’emblée d’orienter vers l’étiologie
Asymétrique
CMH +++
Sarcoïdose
Tumeurs cardiaques
Symétrique
Augmentation de la post charge
Infiltrationmyocardique
HVGPhysiologique
HTARA
Coarctation
AmyloseSarcoïdose
Maladie de Fabry
Cœur du sportif
L’étude de la morphologie de l’HVG, de la fonction VG, de la perfusion du VG, du RT, combinés aux antécédents et aux données échographiques et cliniques,
permet le plus souvent de trouver l’étiologie de l’HVG.

Bibliographie
1. Myerson SG, Bellenger NG, Pennell DJ. Assessment of left ventricular mass by cardiovascular magnetic resonance. Hypertension 2002;39:750-5.
2. Mahrholdt H, Wagner A, Judd RM, Sechtem U, Kim RJ. Delayed enhancement cardiovascular magnetic resonance assessment of non-ischaemic cardiomyopathies. Eur Heart J 2005;26:1461-74.
3. Elliott P, McKenna WJ. Hypertrophic cardiomyopathy. Lancet 2004;363:1881-91.4. Knight CJ. Alcohol septal ablation for obstructive hypertrophic cardiomyopathy. Heart 2006;92:1339-44.5. Maron BJ, McKenna WJ, Danielson GK, et al. American College of Cardiology/European Society of
Cardiology Clinical Expert Consensus Document on Hypertrophic Cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. Eur Heart J 2003;24: 1965–91.
6. Dubourg O, Isnard R, Hagege A et al. Doppler echocardiography in familial hypertrophic cardiomyopathy: the French cooperative study. Echocardiography 1995;12:235-41.
7. Pellicia A, Maron BJ, Spataro A et al. The upper limit of physiologic cardiac hypertrophy in highly trainedelite athletes. N Engl J Med 1991;324:295-301.
8. Rickers C, Wilke NM, Jerosch-Herold M, et al. Utility of cardiac magnetic resonance imaging in the diagnosis of hypertrophic cardiomyopathy. Circulation 2005;112:855– 61.
9. Olivotto I, Maron MS, Autore C et al. Assessment and significance of left ventricular mass by cardiovascular magnetic resonance in hypertrophic cardiomyopathy. J Am Coll Cardiol 2008;52:559-66.
10. Binder J, Ommen SR, Gersh BJ, et al. Echocardiography-guided genetic testing in hypertrophic cardiomyopathy: septal morphological features predict the presence of myofilament mutations. Mayo Clin Proc 2006;81:459–67.

Bibliographie
11. Syed IS, Ommen SR, Breen JF, Tajik AJ. Hypertrophic Cardiomyopathy: Identification of MorphologicalSubtypes by Echocardiography and Cardiac Magnetic Resonance Imaging. J Am Coll Cardiol Img2008;377-379.
12. Dong SJ, MacGregor JH, Crawley AP et al. Left ventricular wall thickness and regional systolic function in patients with hypertrophic cardiomyopathy. A three-dimensional tagged magnetic resonance imaging study. Circulation 1994;90:1200-9.
13. Kramer CM, Reichek N, Ferrari VA et al. Regional heterogeneity of function in hypertrophic cardiomyopathy.Circulation 1994;90:186-94.
14. Maron MS, Olivotto I, Betocchi S et al. Effect of left ventricular outflow tract obstruction on clinicaloutcome in hypertrophic cardiomyopathy. N Engl J Med 2003;348:295-303.
15. Maron MS, Olivotto I, Zenovich AG et al. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation 2006;114:2332-9.
16. Soler R, Rodriguez E, Monserrat L, Mendez C, Martinez C. Magnetic resonance imaging of delayedenhancement in hypertrophic cardiomyopathy: relationship with left ventricular perfusion and contractile function. J Comput Assist Tomogr 2006;30:412-20.
17. Sipola P, Lauerma K, Husso-Saastamoinen M et al. First-pass MR imaging in the assessment of perfusion impairment in patients with hypertrophic cardiomyopathy and the Asp175Asn mutation of the alpha-tropomyosin gene. Radiology 2003;226:129-37.
18. Petersen SE, Jerosch-Herold M, Hudsmith LE et al. Evidence for microvascular dysfunction in hypertrophic cardiomyopathy: new insights from multiparametric magnetic resonance imaging. Circulation 2007;115:2418-25.

Bibliographie
19. Camici P, Crea F. Coronary microvascular dysfunction. N Engl J Med 2007;356:830–840.20. Cecchi F, Olivotto I, Gistri R, Lorenzoni R, Chiriatti G, Camici PG. Coronary microvascular dysfunction
and prognosis in hypertrophic cardiomyopathy. N Engl J Med 2003;349:1027–1035.21. Olivotto I, CecchiF, Gistri R et al. Relevance of coronary microvascular flow impairment to long-term
remodeling and systolic dysfunction in hypertrophic cardiomyopathy. J Am Coll Cardiol 2006;47:1043-8.22. Knaapen P, van Dockum WG, Götte MJ et al. Regional heterogeneity of resting perfusion in hypertrophic
cardiomyopathy is related to delayed contrast enhancement but not to systolic function: a PET and MRI study. J Nucl Cardiol 2006;13:660-7.
23. Sotgia B, Sciagra R, Olivotto I et al. Spatial relationship between coronary microvascular dysfunction and delayed contrast enhancement in patients with hypertrophic cardiomyopathy. J Nucl Med 2008;49:1090-6.
24. Moon JC, McKenna WJ, McCrohon JA, Elliott PM, Smith GC, Pennell DJ, et al. Toward clinical riskassessment in hypertrophic cardiomyopathy with gadolinium cardiovascular magnetic resonance. J Am CollCardiol 2003;41:1561-7.
25. Adabag AS, Maron BJ, Appelbaum E, et al. Occurrence and frequency of arrhythmias in hypertrophic cardiomyopathy in relation to delayed enhancement on cardiovascular magnetic resonance. J Am CollCardiol. 2008;51:1369-74.
26. Moon JC, Reed E, Sheppard MN, et al. The histologic basis of late gadolinium enhancement cardiovascular magnetic resonance in hypertrophic cardiomyopathy. J Am Coll Cardiol 2004;43:2260-4.
27. Debl K, Djavidani B, Buchner S et al. Delayed hyperenhancement in magnetic resonance imaging of left ventricular hypertrophy caused by aortic stenosis and hypertrophic cardiomyopathy: visualisation of focal fibrosis. Heart 2006;92:1447-51.
28. Pujadas S, Carreras F, Arrastio X et al. Detection and quantification of myocardial fibrosis in hypertrophic cardiomyopathy by contrast-enhanced cardiovascular magnetic resonance. Rev Esp Cardiol 2007;60:10-4.

Bibliographie
29. Moon JC, Mogensen J, Elliott PM, et al. Myocardial late gadolinium enhancement cardiovascular magnetic resonance in hypertrophic cardiomyopathy caused by mutations in troponin I. Heart 2005;91:1036-40.
30. Bogaert J, Goldstein M, Tannouri F et al. Original report. Late myocardial enhancement in hypertrophic cardiomyopathy with contrast-enhanced MR imaging. AJR Am J Roentgenol 2003;180:981-5.
31. Choudhury L, Mahrholdt H, Wagner A et al. Myocardial scarring in asymptomatic or mildly symptomaticpatients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2002;40:2156-64.
32. Kim RJ, Judd RM. Gadolinium-enhanced magnetic resonance imaging in hypertrophic cardiomyopathy: in vivo imaging of the pathologic substrate for premature cardiac death? J Am Coll Cardiol 2003;41:1568-72.
33. Nazarian S, Lima JA. Cardiovascular magnetic resonance for risk stratification of arrhythmia in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2008;51:1375-6.
34. Amano Y, Takayama M, Amano M, Kumazaki T. MRI of cardiac morphology and function after percutaneous transluminal septal myocardial ablation for hypertrophic obstructive cardiomyopathy. AJR Am J Roentgenol 2004;182:523-7.
35. Van Dockum WG, ten Cate FJ, ten Berg JM et al. Myocardial infarction after percutaneous transluminal septal myocardial ablation in hypertrophic obstructive cardiomyopathy: evaluation by contrast-enhancedmagnetic resonance imaging. J Am Coll Cardiol 2004;43:27-34.
36. Amano Y, Takayama M, Kumita S, Kumazaki T. MR imaging evaluation of regional, remote, and global effects of percutaneous transluminal septal myocardial ablation in hypertrophic obstructive cardiomyopathy. J Comput Assist Tomogr 2007;31:600-4.
37. van Dockum WG, Beek AM, ten Cate FJ et al. Early onset and progression of left ventricular remodelingafter alcohol septal ablation in hypertrophic obstructive cardiomyopathy. Circulation 2005;111:2503-8.
38. Selvanayagam JB, Hawkins PN, Paul B, Myerson SG, Neubauer S. Evaluation and management of the cardiac amyloidosis. J Am Coll Cardiol 2007;50:2101-10.

Bibliographie
39. Rahman JE, Helou EF, Gelzer-Bell R, et al. Noninvasive diagnosis of biopsy-proven cardiac amyloidosis. J Am Coll Cardiol 2004;43:410–5.
40. Shah KB, Inoue Y, Mehra MR. Amyloidosis and the heart. A comprehensive review. Arch Intern Med 2006;166:1805–13.
41. Hosh W, Bock M, Libicher M et al. MR-relaxometry of myocardial tissue: significant elevation of T1 and T2 relaxation times in cardiac amyloidosis.Invest Radiol 2007;42:636-42.
42. Krombach GA, Hahn C, Tomars M et al. Cardiac amyloidosis: MR imaging findings and T1 quantification, comparison with control subjects. J Magn Reson Imaging 2007;25:1283-7.
43. Fattori R, Rocchi G, Celletti F, Bertaccini P, Rapezzi C, Gavelli G. Contribution of magnetic resonance imaging in the differential diagnosis of cardiac amyloidosis and symetric hypertrophic cardiomyopathy. Am Heart J 1998;136:824-30.
44. Celletti F, Fattori R, Napoli G et al. Assessment of restrictive cardiomyopathy of amyloid or idiopathicetiology by magnetic resonance imaging. Am J Cardiol 1999;83:798-801.
45. van den Driesen RI, Slaughter RE, Strugnell WE. MR findings in cardiac amyloidosis. Am J Roentgenol2006;186:1682-5.
46. Sharma PP, Payvar S, Litovsky SH. Histomorphometric analysis of intramyocardial vessels in primary and senile amyloidosis: epicardium versus endocardium. Cardiovasc Pathol 2008;17:65-71.
47. Maceira AM, Joshi J, Prasad SK, et al. Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation 2005;111:122–4.
48. Vogelsberg H, Mahrholdt H, Deluigi CC et al. Cardiovascular magnetic resonance in clinically suspectedcardiac amyloidosis: noninvasive imaging compared to endomyocardial biopsy. J Am Coll Cardiol 2008;51:1022-30.

Bibliographie
49. Perugini E, Rapezzi C, Piva T, et al. Non-invasive evaluation of the myocardial substrate of cardiac amyloidosis by gadolinium cardiac magnetic resonance. Heart 2006;92:343–9.
50. Otto CM. Valvular aortic stenosis: disease severity and timing of intervention. J Am Coll Cardiol 2006;47:2141-51.
51. Braunwald’s heart disease: a textbook of cardiovascular medicine. Libby P, Bonow RO, Mann DL, ZipesDP. 8th ed. 1625-1635.
52. John AS, Dill T, Brandt RR et al. Magnetic resonance to assess the aortic valve area in aortic stenosis: how does it compare to current diagnostic standards? J Am Coll Cardiol 2003;42:519-26.
53. Kupfahl C, Honold M, Meinhardt G et al. Evaluation of aortic stenosis by cardiovascular magnetic resonance: comparison with established routine clinical techniques. Heart 2004;90:893-901.
54. Reant P, Lederlin M, Lafitte S et al. Absolute assessment of aortic valve stenosis by planimetry using cardiovascular magnetic resonance imaging: comparison with transesophageal echocardiography, transthoracic echocardiography, and cardiac catheterisation. Eur J Radiol 2006;59:276-83.
55. Debl K, Djavidani B, Seitz J et al. Planimetry of aortic valve area in aortic stenosis by magnetic resonance imaging. Invest Radiol 2005;40:631-6.
56. Caruthers SD, Lin SJ, Brown P et al. Practical value of cardiac magnetic resonance imaging for clinicalquantification of aortic valve stenosis: comparison with echocardiography. Circulation 2003;108:2236-43.
57. Pouleur AC, le Polain de Warroux JB, Pasquet A et al. Planimetric and continuity equation assessment of aortic valve area: Head to head comparison between cardiac magnetic resonance and echocardiography. J Magn Reson Imaging 2007;26:1436-43.
58. Tanaka K, Makaryus AN, Wolff SD. Correlation of aortic valve area obtained by the velocity-encoded phase contrast continuity method to direct planimetry using cardiovascular magnetic resonance. JCardiovasc Magn Reson 2007;9:788-805.

Bibliographie
59. Debl K, Djavidani B, Buchner S et al. Delayed hyperenhancement in magnetic resonance imaging of left ventricular hypertrophy caused by aortic stenosis and hypertrophic cardiomyopathy: visualisation of focal fibrosis. Heart 2006;92:1447-51.
60. Hein S, Arnon E, Kostin S, et al. Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: structural deterioration and compensatory mechanisms. Circulation 2003;107:984–91.
61. Krayenbuehl HP, Hess OM, Monrad ES, et al. Left ventricular myocardial structure in aortic valve disease before, intermediate, and late after aortic valve replacement. Circulation 1989;79:744–55.
62. Maron BJ, Shirani J, Poliac LC, Mathenge R, Roberts WC, Mueller FO. Sudden death in youngcompetitive athletes: clinical, demographic, and pathological profiles. JAMA 1996;276:199 –204
63. Maron BJ. Sudden death in young athletes. N Engl J Med 2003;349:1064–75.64. Maron BJ, Thompson PD, Ackerman MJ et al. Recommendations and considerations related to
preparticipation screening for cardiovascular abnormalities in competitive athletes: 2007 update: a scientificstatement from the American Heart Association Council on Nutrition, Physical Activity, and Metabolism: endorsed by the American College of Cardiology Foundation. Circulation 2007;115:1643-55
65. Corrado D, Basso C, Schiavon M, Thiene G. Screening for hypertrophic cardiomyopathy in young athletes. N Engl J Med 1998;339:364-9.
66. Basavarajaiah S, Boraita A, Whyte G et al. Ethnic differences in left ventricular remodeling in highly-trained athletes relevance to differentiating physiologic left ventricular hypertrophy from hypertrophic cardiomyopathy. J Am Coll Cardiol 2008;51:2256-62.
67. Pelliccia A, Fagard R, Bjørnstad HH et al. A European Society of Cardiology consensus document: recommendations for competitive sports participation in athletes with cardiovascular disease. Eur Heart J. 2005;26:1422–1445.

Bibliographie
68. Pelliccia A, Di Paolo FM, Corrado D, et al. Evidence for efficacy of the Italian national pre-participation screening programme for identification of hypertrophic cardiomyopathy in competitive athletes. Eur Heart J 2006;27:2196 –200.
69. Corrado D, Basso C, Pavei A et al. Trends in sudden cardiovascular death in young competitive athletesafter implementation of a preparticipation screening program. JAMA 2006;296:1593-601.
70. Maron BJ, Pelliccia A. The heart of trained athletes: cardiac remodeling and the risks of Sports, includingsudden death. Circulation. 2006;114:1633-1644.
71. Savage DD, Seides SF, Clark CE et al. Electrocardiographic findings in patients with obstructive and non obstructive hypertrophic cardiomyopathy. Circulation 1978;58:402– 8.
72. Maron BJ, Wolfson JK, Ciro E, Spirito P. Relation of electrocardiographic abnormalities and patterns of hypertrophy identified by 2-dimensional echocardiography in patients with hypertrophic cardiomyopathy. Am J Cardiol 1983;51:189.
73. Sharma S, Whyte G, Elliott P, et al. Electrocardiographic changes in 1000 highly trained junior eliteathletes. Br J Sports Med 1999;33:319–24.
74. Corrado D, McKenna WJ. Appropriate interpretation of the athlete’s electrocardiogram saves lives as wellas money. Eur Heart J 2007;28:1920 –2.
75. Pelliccia A, Di Paolo FM, Quattrini FM et al. Outcomes in Athletes with Marked ECG RepolarizationAbnormalities. N Engl J Med 2008;358:152-61.
76. Pelliccia A, Maron BJ, Culasso F et al. Clinical significance of abnormal electrocardiographic patterns in trained athletes. Circulation 2000;102:278-84.
77. Klues HG, Roberts WC, Maron BJ. Morphologic determinants of echocardiographic patterns of mitral valve systolic anterior motion in obstructive hypertrophic cardiomyopathy. Circulation 1993;87:1570–9.
78. Lewis JF, Spirito P, Pellicia A, Maron BJ. Usefulness of Doppler echocardiographic assessment of diastolic filling in distinguishing “athlete’s heart” from hypertrophic cardiomyopathy. Br Heart J 1992;68:296 –300.

Bibliographie
79. Maron BJ, Pellicia A, Spirito P. Cardiac disease in young trained athletes. Insights into methods for distinguishing athletes heart from structural heart disease, with particular emphasis on hypertrophic cardiomyopathy. Circulation 1995;91:1596–601.
80. Sharma S, Elliott PM, Whyte G, et al. Utility of metabolic exercise testing in distinguishing hypertrophic cardiomyopathy from physiologic left ventricular hypertrophy in athletes. J Am Coll Cardiol 2000;36:864–70.
81. Maron BJ, Pelliccia A, Spataro A, Granata M. Reduction in LV wall thickness after deconditioning in highly trained Olympic athletes. Br Heart J 1993;69:125– 8.
82. Basavarajaiah S, Wilson M, Junagde S, Jackson G, Whyte G, Sharma S. Physiological LV hypertrophy or hypertrophic cardiomyopathy in an elite adolescent athlete: role of detraining in resolving the clinicaldilemma. Br J Sports Med 2006;40:727–9.
83. Ehsani AA, Hagberg JM, Hickson RC. Rapid changes in LV dimensions and mass in response to physicalconditioning and deconditioning. Am J Cardiol 1978;42:52– 6.
84. Pelliccia A, Maron BJ, De Luca R et al. Remodeling of left ventricular hypertrophy in elite athletes after long-term deconditioning. Circulation 2002;105:944-9.
85. Moon JC, Fisher NG, McKenna WJ, Pennell DJ. Detection of apical hypertrophic cardiomyopathy by magnetic resonance in patients with non-diagnostic echocardiography. Heart 2004;90:645–9.
86. Petersen SE, Selvanayagam JB, Francis JM et al. Differentiation of athlete's heart from pathological formsof cardiac hypertrophy by means of geometric indices derived from cardiovascular magnetic resonance. J Cardiovasc Magn Reson 2005;7:551-8.
87. Pelliccia A, Maron BJ, Di Paolo FM, et al. Prevalence and clinical significance of left atrial remodeling in competitive athletes. J Am Coll Cardiol 2005;46:690–6.
88. Perry A, Vuitch F. Causes of death in patients with sarcoidosis. A morphologic study of 38 autopsies with clinicopathologic correlations. Arch Pathol Lab Med 1995;119:167–72.

Bibliographie
89. Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. N Engl J Med 2007;357:2153-65.90. Uemura A, Morimoto S, Hiramitsu S, Kato Y, Ito T, Hishida H. Histologic diagnostic rate of cardiac
sarcoidosis: evaluation of endomyocardial biopsies. Am Heart J 1999;138:299–302.91. Mehta D, Libitz SA, Frankel Z et al. Cardiac involvement in patients with sarcoidosis: diagnostic and
prognostic value of outpatient testing. Chest 2008;133:1426-35.92. Smedena JP, Snoep G, van Kroonenburgh MP et al. Evaluation of the accuracy of gadolinium-enhanced
cardiovascular magnetic resonance in the diagnosis of cardiac sarcoidosis. J Am Coll Cardiol 2005;45:1683-90.
93. Vignaux O, Dhote R, Duboc D et al. Clinical significance of myocardial magnetic resonance abnormalitiesin patients with sarcoidosis: a 1-year follow-up study. Chest 2002;122:1895-901.
94. Vignaux O, Dhote R, Duboc D et al. Detection of myocardial involvement in patients with sarcoidosisapplying T2-weighted, contrast-enhanced, and cine magnetic resonance imaging: initial results of a prospective study. J Comput Assist Tomogr 2002;26:762-7.
95. Vignaux O. Cardiac Sarcoidosis: Spectrum of MRI Features. Am J Roentgenenol 2005;184:249–254.96. Ichinose A, Otani H, Oikawa M et al. MRI of cardiac sarcoidosis: basal and subepicardial localization of
myocardial lesions and their effect on left ventricular function. Am J Roentgenol 2008;191:862-9.97. Yoshida Y, Morimoto S. Hiramitsu, Tsuboi N, Hirayama H, Itoh T. Incidence of cardiac sarcoidosis in
japanese patients with high-degree atrioventricular block. Am Heart J 1997;134:382–6.98. Chapelon-Abric C, de Zuttere D, Duhaut P et al. Cardiac sarcoidosis: a retrospective study in 41 cases.
Medicine (Baltimore) 2004;83:315–24.99. Clarke JT. Narrative review: Fabry disease. Ann Intern Med 2007;146:425-33.
100. Linhart A, Elliott PM. The heart in Anderson-Fabry disease and other lysosomal storage disorders. Heart 2007;93:528-35.

Bibliographie
101. Schiffmann R, Kopp JB, Austin HA 3rd, et al. Enzyme replacement therapy in Fabry disease: a randomizedcontrolled trial. JAMA 2001;285: 2743–49.
102. Eng CM, Guffon N, Wilcox WR, et al. Safety and efficacy of recombinant human -galactosidase A replacement in Fabry disease. N Engl J Med 2001; 345: 9–16.
103. Pastores GM, Thadhani R. Enzyme-replacement therapy for Anderson–Fabry disease. Lancet 2001; 358:601–603.
104. Hughes DA, Elliott PM, Shah J, et al. Effects of enzyme replacement therapy on the cardiomyopathy of Anderson–Fabry disease: a randomized, doubleblind, placebo-controlled clinical trial of agalsidasealfa. Heart 2008;94:153–8.
105. Sachdev B, Takenaka T, Teraguchi H, et al. Prevalence of Anderson-Fabry disease in male patients with late onset hypertrophic cardiomyopathy. Circulation 2002;105:1407–11.
106. Chimenti C, Pieroni M, Morgante E, et al. Prevalence of Fabry disease in female patients with late-onsethypertrophic cardiomyopathy. Circulation 2004;110:1047–53.
107. Monserrat L, Gimeno-Blanes JR, Marin F et al. Prevalence of fabry disease in a cohort of 508 unrelatedpatients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2007;50:2399-403.
108. Linhart A, Kampmann C, Zamorano JL et al. Cardiac manifestations of Anderson-Fabry disease: resultsfrom the international Fabry outcome survey. Eur Heart J 2007;28:1228-35.
109. Matsui S, Murakami E, Takekoshi N, Nakatou H, Enyama H, Takeda F. Myocardial tissue characterizationby magnetic resonance imaging in Fabry’s disease. Am Heart J 1989; 117:472–474.
110. Imbriaco M, Spinelli L, Cuocolo A et al. MRI characterization of myocardial tissue in patients with Fabry's disease. AJR Am J Roentgenol 2007;188:850-3.
111. Lidove O, Klein I, Lelièvre JD, Lavallée P, Serfaty JM, Dupuis E, Papo T, Laissy JP. Imaging features of Fabry disease.AJR Am J Roentgenol 2006;186:1184-91.

Bibliographie
112. Moon JC, Sachdev B, Elkington AG, et al. Gadolinium enhanced cardiovascular magnetic resonance in Anderson–Fabry disease: evidence for a disease specific abnormality of the myocardial interstitium. Eur Heart J 2003; 24:2151–2155.
113. Drazner MH, Dries DL, Peshock RM et al. Left ventricular hypertrophy is more prevalent in blacks thanwhites in the general population: the Dallas Heart Study. Hypertension 2005;46:124-9.
114. Gradman AH, Alfayoumi F. From left ventricular hypertrophy to congestive heart failure: management of hypertensive heart disease. Prog Cardiovasc Dis 2006;48:326-41.
115. Rosen BD, Edvardsen T, Lai S et al. Left ventricular concentric remodeling is associated with decreasedglobal and regional systolic function: the Multi-Ethnic Study of Atherosclerosis. Circulation 2005;112:984-91.
116. Drazner MH. The transition from hypertrophy to failure: how certain are we? Circulation 2005;936-8.117. Andersen K, Hennersdorf M, Cohnen M et al. Myocardial delayed contrast enhancement in patients with
arterial hypertension: Initial results of cardiac MRI. Eur J Radiol 2008 Apr 21 [Epub ahead of print].118. Sparrow PJ, Kurian JB, Jones TR, Sivananthan MU. MR imaging of cardiac tumors. Radiographics
2005;25:1255-76.119. Mollet NR, Dymarkowski S, Volders W, et al. Visualization of ventricular thrombi with contrast-enhanced
magnetic resonance imaging in patients with ischemic heart disease. Circulation 2002;106:2873-6.120. Srichai MB, Junor C, Rodriguez LL, et al. Clinical, imaging, and pathologic characteristics of left
ventricular thrombus: a comparison of contrast enhanced magnetic resonance imaging, transthoracicechocardiography and transesophageal echocardiography with surgical or pathological validation. Am Heart J 2006;152:75-84.
121. Weinsaft JW, Kim HW, Shah DJ, et al. Detection of left ventricular thrombus by delayed-enhancement cardiovascular magnetic resonance prevalence and markers in patients with systolic dysfunction. J Am CollCardiol 2008;52:148-57.

Bibliographie
122. Jenni R, Oechslin EN, van der Loo B. Isolated ventricular non-compaction of the myocardium in adults. Heart 2007;93:11-5.
123. Jenni R, Oechslin E, Schneider J, Attenhofer Jost C, Kaufmann PA. Echocardiographic and pathoanatomical characteristics of isolated left ventricular non-compaction: a step towards classification as a distinct cardiomyopathy. Heart 2001;86:666–671.
124. Petersen SE, Selvanayagam JB, Wiesmann F et al. Left ventricular non-compaction: insights fromcardiovascular magnetic resonance imaging. J Am Coll Cardiol 2005;46:101-5.