Ecole Doctorale Gay-Lussac 2009-2010 Techniques de...
93
1 Caractérisation des catalyseurs solides. Adsorption de molécules-sondes suivie par spectroscopie infrarouge Sébastien Laforge LACCO, UMR 6503 CNRS – Université de Poitiers Ecole Doctorale Gay-Lussac 2009-2010 Techniques de caractérisation
Transcript of Ecole Doctorale Gay-Lussac 2009-2010 Techniques de...

1
Caractérisation des catalyseurs solides. Adsorption de molécules-sondes suivie par
spectroscopie infrarouge
Sébastien Laforge
LACCO, UMR 6503 CNRS –
Université
de Poitiers
Ecole Doctorale Gay-Lussac
2009-2010
Techniques de caractérisation

2
1ère partie :
Généralités sur la spectroscopie d’absorption infrarouge.

3
1. Niveaux d’énergie, transitions et spectroscopie
Théorie des quanta de Planck
(1900): la lumière est constituée de corpuscules appelés photons, dont l’énergie est quantifiée et s’exprime par:
Où
h est la constante de Planck (= 6,626.10−34
J s), la fréquence du rayonnement (en Hz), sa longueur d’onde, son nombre d’ondes et c la vitesse de la lumière (~ 3.108
m.s−1).
chchhE
Cette relation entre la fréquence d’un rayonnement et son énergie est le fondement de toutes les spectroscopies et en particulier des spectroscopies vibrationnelles.
1.1. Caractère corpusculaire de la lumière
4
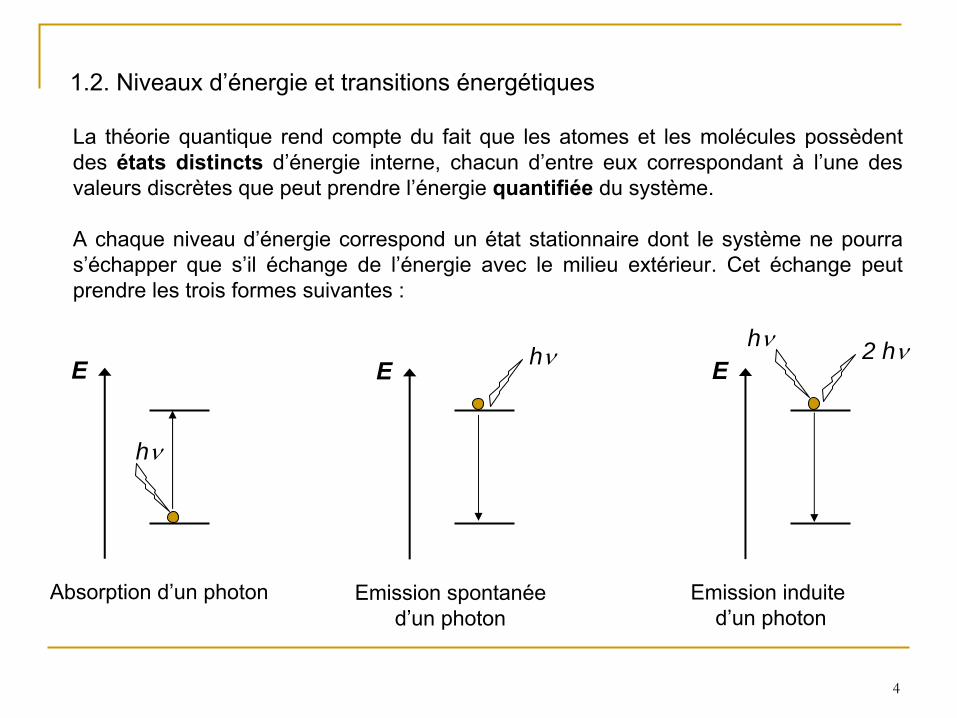
1.2. Niveaux d’énergie et transitions énergétiques
La théorie quantique rend compte du fait que les atomes et les molécules possèdent des états distincts
d’énergie interne, chacun d’entre eux correspondant à
l’une des valeurs discrètes que peut prendre l’énergie quantifiée du système.
A chaque niveau d’énergie correspond un état stationnaire dont le système ne pourra s’échapper que s’il échange de l’énergie avec le milieu extérieur. Cet échange peut prendre les trois formes suivantes :
E
h
Absorption d’un photon
E h
Emission spontanéed’un photon
E
Emission induite d’un photon
2 hh

5
1.3. Spectroscopies atomiques et moléculaires
Comme nous venons de le voir, un atome peut absorber ou émettre un rayonnement comme résultat d’une transition entre différents états énergétiques. Son énergie «
électronique
»
Eel peut ainsi être modifiée.
Il en est de même pour les molécules, dont l’énergie peut varier selon deux modes qui ne sont pas envisageables pour l’atome seul :
• la diminution ou l’augmentation de leur énergie de vibration Evib
• la diminution ou l’augmentation de leur énergie de rotation Erot
Comme l’énergie «
électronique
»
Eel , Evib et Erot sont quantifiées, mais les ordres de grandeurs des écarts entre deux niveaux d’énergie successifs sont très différents :
Eel = 25 kJ.mol−1; Evib = 1 kJ.mol−1
et Erot = 0,0025 kJ.mol−1
On peut donc représenter les différents «
niveaux
»
d’énergie des molécules de la manière suivante :

6
E
Transitionsélectroniques
Transitionsvibrationnelles
0
1
2
123
Transitionsrotationnelles
Si l’on considère le processus d’absorption d’un rayonnement, la nature des transitions observées dépendra donc de la longueur d’onde du photon absorbé:
chchhE
7
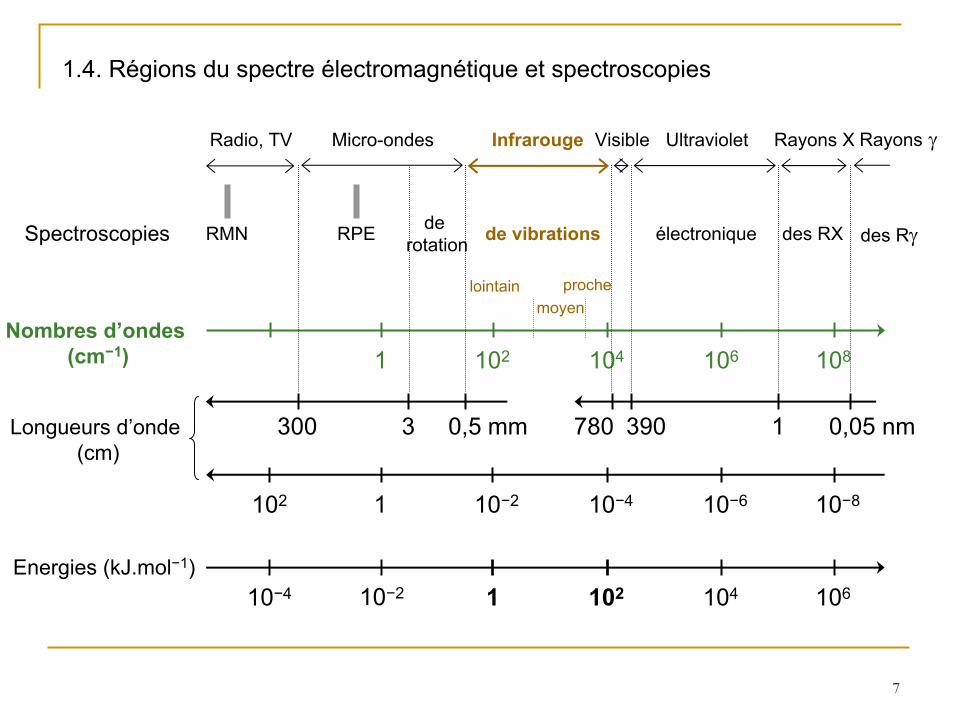
1 102 104 106 108Nombres d’ondes
(cm−1)
1 10−2 10−4 10−6 10−8
Longueurs d’onde (cm)
102
300 3 0,5 mm 390780 1 0,05 nm
Micro-ondes Visible Ultraviolet Rayons X Rayons
Radio, TV Infrarouge
Spectroscopies RMN RPE de rotation de vibrations électronique des RX des R
lointain proche
10−4 1 102 104 10610−2Energies (kJ.mol−1)
1.4. Régions du spectre électromagnétique et spectroscopies
moyen

8
1.5. Transitions énergétiques et peuplement des niveaux d’énergie
Pour qu’un état donné
soit le point de départ d’une transition, encore faut-il que des molécules soient présentes dans cet état.
Le peuplement des différents états énergétiques peut être estimé
à
partir de la formule de Boltzmann, qui permet d’estimer, à
une température T le rapport des populations de deux niveaux i et j :
T6952,0exp
gg
Tkchexp
gg
TkEE
expgg
NN
i
j
Bi
j
B
ij
i
j
i
j
où
gi et gj représentent les dégénérescences des états i et j.

9
A 298 K et si l’on considère que les niveaux ne sont pas dégénérés (g= 1), on obtient :
01 NN
2.10−3(cm−1) 20 200 2000
0,9999 0,91 0,38 0,00007
2.104
2.10−42
Les états «
excités
»
thermiquement sont quasiment vides en spectroscopie électronique et en spectroscopie vibrationnelle pour les vibrations de fréquence élevée. Si l’on effectue les analyses spectroscopiques à
température ambiante, les seules transitions observées auront le niveau fondamental pour origine.
Par contre, un certain nombre de niveaux excités d’énergie rotationnelle sont peuplés. On pourra donc observer un grand nombre de transitions rotationnelles, y compris entre des niveaux excités.
Rot. Vib.Type Elect.

10
La conséquence est que, lors de l’absorption d’un rayonnement infrarouge polychromatique, les transitions énergétiques entre les niveaux de vibration s’accompagneront d’un grand nombre de transitions rotationnelles. Par conséquent, on observera des «
bandes
»
d’absorption, plus ou moins larges, plutôt que des raies discrètes.
11
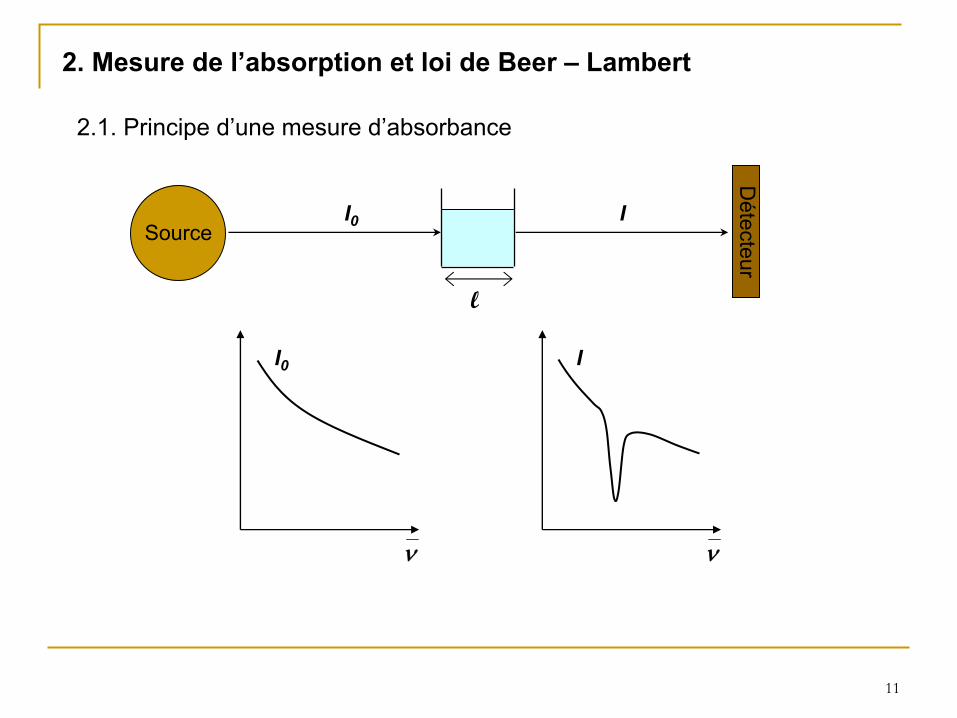
2. Mesure de l’absorption et loi de Beer –
Lambert
I0
I0 I
I
2.1. Principe d’une mesure d’absorbance
l
SourceD
étecteur

12
2.2. Loi de Beer -
Lambert
Lorsqu’un rayonnement monochromatique (de nombre d’ondes ) traverse une solution formée d’un soluté
absorbant dans un solvant transparent, la diminution de son intensité
est proportionnelle à
l’épaisseur l
de la solution et à
la concentration c du soluté.
l cexpII 0
où
est le coefficient d’absorption molaire
(mol−1
cm−2) du soluté
au nombre d’ondes
.
Le rapport I / I0 , exprimé
en %,
est appelé
transmission
(T ).
L’absorbance
A (ou densité
optique D) est définie par : l
c
T1logA 10
où
est le coefficient d’extinction molaire
(mol−1
cm−2) du soluté
au nombre d’ondes
.
3,2 Remarque :
13
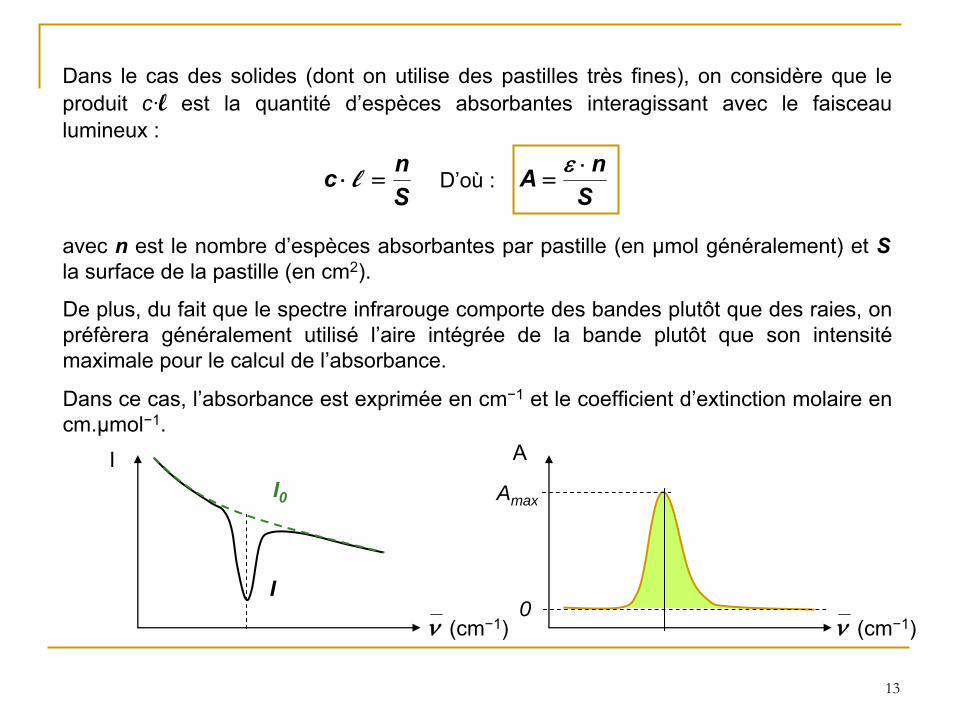
Dans le cas des solides (dont on utilise des pastilles très fines), on considère que le produit c·l
est la quantité
d’espèces absorbantes interagissant avec le faisceau lumineux :
Snc l
avec n
est le nombre d’espèces absorbantes par pastille (en µmol généralement) et S
la surface de la pastille (en
cm2).
D’où
:S
nA
De plus, du fait que le spectre infrarouge comporte des bandes plutôt que des raies, on préfèrera généralement utilisé
l’aire intégrée de la bande plutôt que son intensité
maximale pour le calcul de l’absorbance.
Dans ce cas, l’absorbance est exprimée en cm−1
et le coefficient d’extinction molaire en cm.µmol−1.
I
I0
(cm−1)
I A
(cm−1)
Amax
0

14
3. Modes normaux de vibration des molécules polyatomiques
Un mode normal de vibration est un mouvement indépendant et synchrone
d’atomes ou de groupes d’atomes qui peut être excité
sans entraîner l’excitation d’un autre mode normal.
Le nombre de degrés de liberté
que possède une molécule est la somme des degrés de liberté
des différents atomes qu
’elle contient. Or, chaque atome en possède trois, correspondant aux coordonnées cartésiennes (x, y et z) nécessaires pour décrire sa position dans l’espace. Une molécule comportant N atomes possèdent donc 3 N
degrés de libertés.Trois de ces degrés de liberté
décrivent la translation de la molécule.Dans les molécules non linéaires, trois autres degrés de liberté
correspondent à
la rotation de la molécule sur elle-même. Il n’y en a que deux pour les molécules linéaires.
En conséquence, pour une molécule non linéaire comportant N atomes, il existe (3 N −
6) degrés de liberté
de vibration et donc (3 N −
6) modes de vibration indépendants. Il n’y en a que (3 N −
5) si la molécule est linéaire.
3.1. Définition

15
Parmi les différents modes de vibration on distinguera :
Elongation symétrique«
Symmetrical stretching
»Elongation asymétrique
«
Asymmetrical stretching
»
Cisaillement« Scissoring »
Balancement horsdu plan «
Wagging
»Torsion
« Twisting »Balancement dans le plan «
Rocking
»
• Les modes d’élongation , dans lesquels la longueur des liaisons est modifiée.
•
Les modes de déformation angulaire , dans lesquels les angles entre les liaisons sont modifiés.

16
3.2. Exemples
1) CO2
: (3
N −
5) = 4Elongation symétrique Elongation asymétrique Cisaillement
(s
CO) (1388 cm−1) (as
CO) (2349 cm−1) (s
CO2 ) (667 cm−1)
2) H2
O : (3
N −
6) = 3
s
OH) (3652 cm−1) (s
H2
O) (1595 cm−1) (as
OH) (3756 cm−1)
Elongation(+ déformation)
Elongation(+ déformation)
Cisaillement(+ élongation)
3) Naphtalène C10
H8
: (3
N −
6) = 48

17
3.3. Remarque
Pour les molécules CO2
et H2
O, les nombres d’ondes associés aux vibrations de déformation sont nettement plus petits que ceux relatifs aux modes
d’élongation.

18
3.3. Modes de vibration et nombre d’ondes
En première approximation, on peut considérer que chaque mode normal se comporte comme un oscillateur harmonique (deux masses reliées par un ressort) indépendant. Le nombre d’ondes de la vibration peut alors être estimé
à
partir de la loi de Hooke :
mk
c21
où
k est la constante de force relative au mode de vibration considéré
et m la masse apparente. Cette dernière est une mesure de la masse «
ballottée
»
au cours de la vibration. C’est en général une fonction complexe des masses des atomes en mouvement.
Par exemple, dans la vibration d’élongation symétrique de CO2
seuls les atomes d’oxygène se déplacent. La masse apparente ne dépend que d’eux. Par contre, lors de l’élongation asymétrique, les trois atomes entrent en mouvement, de sorte qu’ils contribuent tous à
la masse apparente.

19
On peut facilement mettre en évidence la relation entre la force d’une liaison et le nombre d’ondes de la vibration d’élongation associée à
cette liaison en comparant les valeurs «
théoriques
»
suivantes :
C C C C C C
2150 1650 1200(cm−1)
HC
3300 3100 2900(cm−1)
HC HC
HC
3000 1200 800(cm−1)
CC ClC
1100
OC
De même, pour l’influence des masses atomiques :

20
Ainsi, chaque mode normal de vibration devrait se traduire par l’apparition d’une bande, à
un nombre d’ondes donné. Ce n’est généralement pas le cas.
Par exemple, dans le cas de CO2
(en phase gaz), seules deux bandes sont observées en spectroscopie IR, à
2349 et 667 cm−1, alors que la molécule possède
4 modes normaux de vibration:
Elongation symétrique Elongation asymétrique Déformation1
(1388 cm−1) 3
(2349 cm−1) 2
(667 cm−1)
Ceci s’explique par le fait que:
1) les deux modes de déformation sont strictement équivalents et conduisent à
la même valeur du nombre d’ondes (ils forment un mode doublement dégénéré);
2) Le mode d’élongation symétrique est inactif en infrarouge.

21
3.4. Modes actifs en spectroscopie IR
La règle de sélection générale pour l’activité
infrarouge stipule que le mouvement correspondant à un mode normal de la molécule doit s’accompagner d’une variation de son moment dipolaire.
Un mode normal de vibration qui ne respecte pas cette règle est dit inactif en infrarouge. Il n’y aura donc pas de bande d’absorption dans le spectre infrarouge au nombre d’ondes correspondant à
ce mode.
C’est le cas du mode d’élongation symétrique de CO2
:
1
(1388 cm−1)
Ceci explique pourquoi les molécules diatomiques homonucléaires (N2
, O2
, H2
, Cl2
, ...), pour lesquelles le seul mode normal de vibration est une élongation symétrique de la liaison) ne sont observables en spectroscopie IR.

22
4. Appareillage
Aujourd’hui, la majeure partie des spectromètres infrarouge utilisés sont des spectromètres à
transformée de Fourier (FT-IR).
Cette technologie permet, entre autre, d’utiliser une gamme complète de radiations couvrant le domaine du moyen (ou du proche) infrarouge et donc d’obtenir très rapidement un spectre complet.
De plus, le traitement numérique du signal rend aisé
le traitement des spectres et permet d’obtenir d’excellents spectres à
partir de très petits échantillons.
23
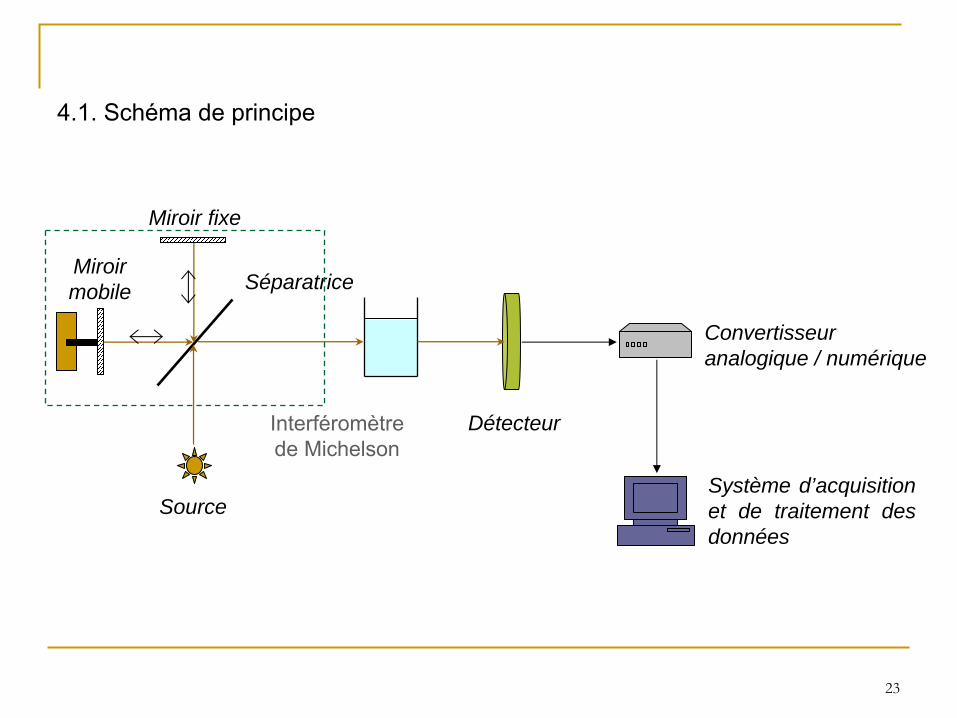
4.1. Schéma de principe
Source
Miroirmobile
Miroir fixe
Séparatrice
Détecteur
Convertisseuranalogique / numérique
Système d’acquisition et de traitement des données
Interféromètre de Michelson

24
La radiation infrarouge est produite par chauffage de 1000 à
1800°C d’une source qui est souvent un filament de Nernst (alliage d’oxydes de Zr, Th et Ce) ou un globar (barreau de carbure de silicium).
La séparatrice de l’interféromètre est en KBr pour l’
infrarouge moyen.
Les détecteurs les plus courant en infrarouge moyen sont :
•
soit de type quantique (comptage des photons): MCT (tellurure de mercure et de cadmium);
•
soit de type thermique (sensible à
la température dégagée par une cible): DTGS (sulfate de triglycine deutérée).
4.2. Instrumentation
25
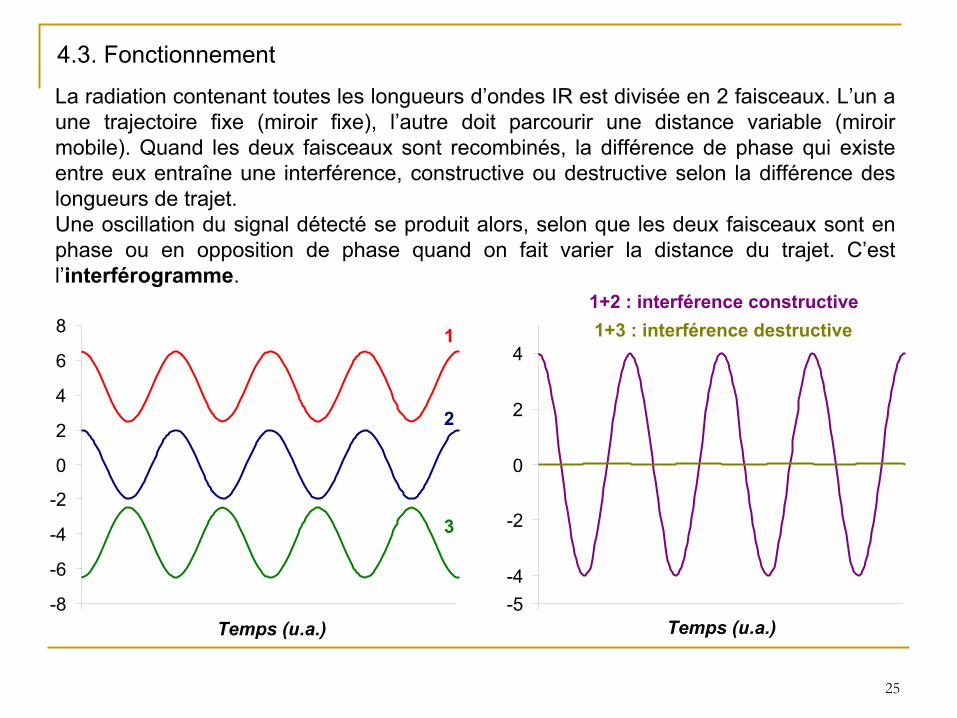
La radiation contenant toutes les longueurs d’ondes IR est divisée en 2 faisceaux. L’un a une trajectoire fixe (miroir fixe), l’autre doit parcourir une distance variable (miroir
mobile). Quand les deux faisceaux sont recombinés, la différence de phase qui existe entre eux entraîne une interférence, constructive ou destructive selon la différence des longueurs de trajet. Une oscillation du signal détecté
se produit alors, selon que les deux faisceaux sont en phase ou en opposition de phase quand on fait varier la distance
du trajet. C’est l’interférogramme.
4.3. Fonctionnement
-8
-6
-4
-2
0
2
4
6
8 1
2
3
-5-4
-2
0
2
4
1+2 : interférence constructive1+3 : interférence destructive
Temps (u.a.) Temps (u.a.)
26
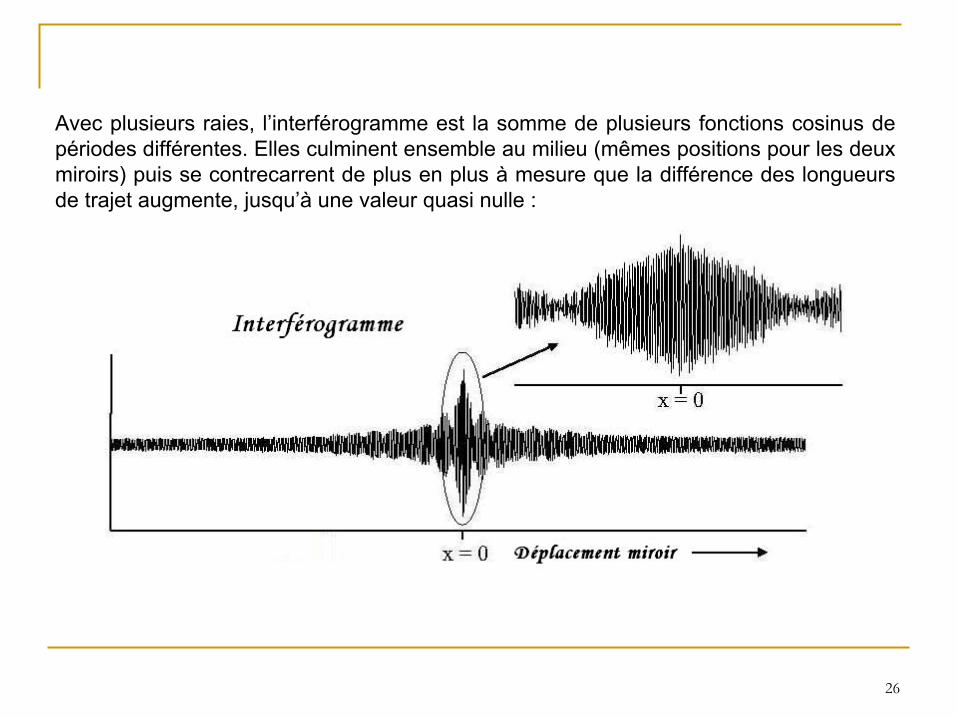
Avec plusieurs raies, l’interférogramme est la somme de plusieurs fonctions cosinus de périodes différentes. Elles culminent ensemble au milieu (mêmes positions pour
les deux miroirs) puis se contrecarrent de plus en plus à
mesure que la différence des longueurs de trajet augmente, jusqu’à
une valeur quasi nulle :
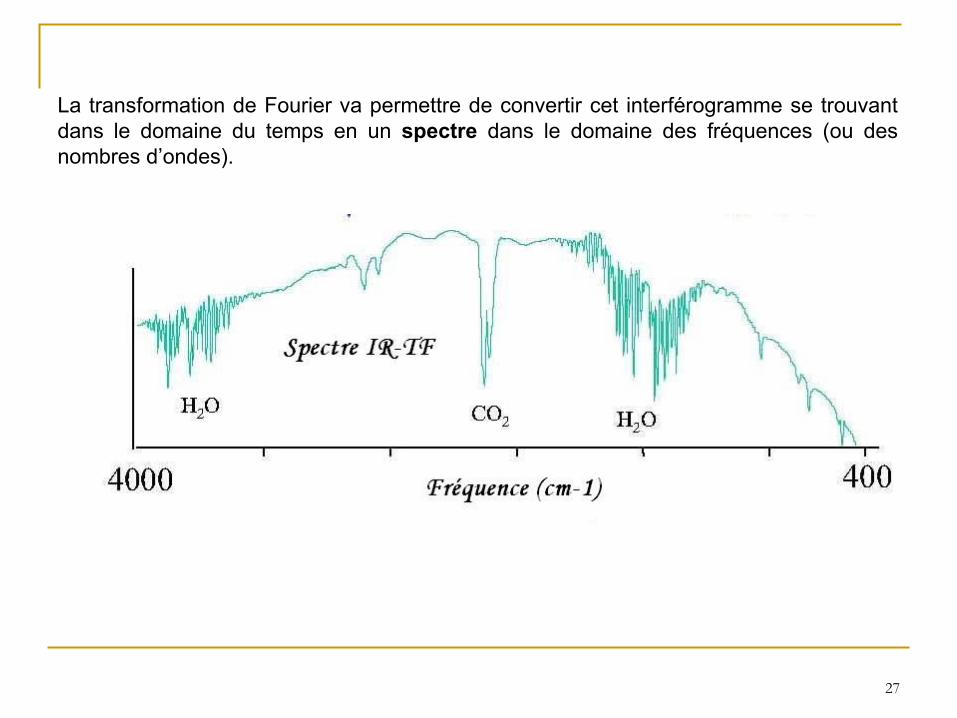
27
La transformation de Fourier va permettre de convertir cet interférogramme se trouvant dans le domaine du temps en un spectre
dans le domaine des fréquences (ou des nombres d’ondes).

28

29
2nde partie :
Applications de la spectroscopie d’absorption infrarouge en chimie.
• Identification des composés organiques
•
Caractérisation des catalyseurs solides par adsorption de molécules- sondes
1. Catalyseurs métalliques supportés (Pt, Pd)
2. Catalyseurs zéolithiques

30
A/
Identification des composés organiques
31
Ces molécules sont généralement analysées en phase liquide, diluées dans un solvant. Bien évidemment, le solvant choisi ne devra pas absorber le rayonnement infrarouge dans le même domaine de nombre d’ondes que le soluté.
Les solvants les plus courants sont le tétrachlorure de carbone (CCl4
), qui est pratiquement dépourvu d’absorption au dessus de 1333 cm−1) et le disulfure de
carbone CS2
.
Les solutions sont introduites dans des cellules munies de fenêtres transparentes aux rayonnements infrarouges.

32
Les solutions sont introduites dans des cellules munies de fenêtres transparentes aux rayonnements infrarouges. Les matériaux les plus couramment utilisés sont présentés ci-dessous :
Fenêtre
Domaine spectral (cm-1)
Sensibilité
à
l'eau
Coût
CaF2
10000 –
1000
Peu soluble
Moyen
NaCl
10000 –
500
Soluble
Moyen
KBr
10000 –
300
Très soluble
Moyen
CsI
10000 –
170
Très soluble
Très cher
ZnSe
10000 -
500
Insoluble
Très cher
On peut également travailler à
partir du composé
liquide pur, par préparation d’un film de quelques dizaines de microns d’épaisseur entre deux fenêtres, généralement en NaCl.

33
Une analyse précise des vibrations d’une molécule complexe n’est pas
envisageable. Dès lors, le spectre IR doit être interprété
à
partir de comparaisons empiriques de spectres et par extrapolations sur des molécules plus simples.
Les domaines de nombres d’ondes dans lesquels vibrent les différents types de liaisons au sein des molécules organiques sont généralement disponibles sous la forme de tables :
Vibration
(cm−1)
(C-H)
2850 -
2960
(C-H)
1340 -
1465
(C-C),
(C-C)
700 -
1250
(C=C)
1620 -
1680
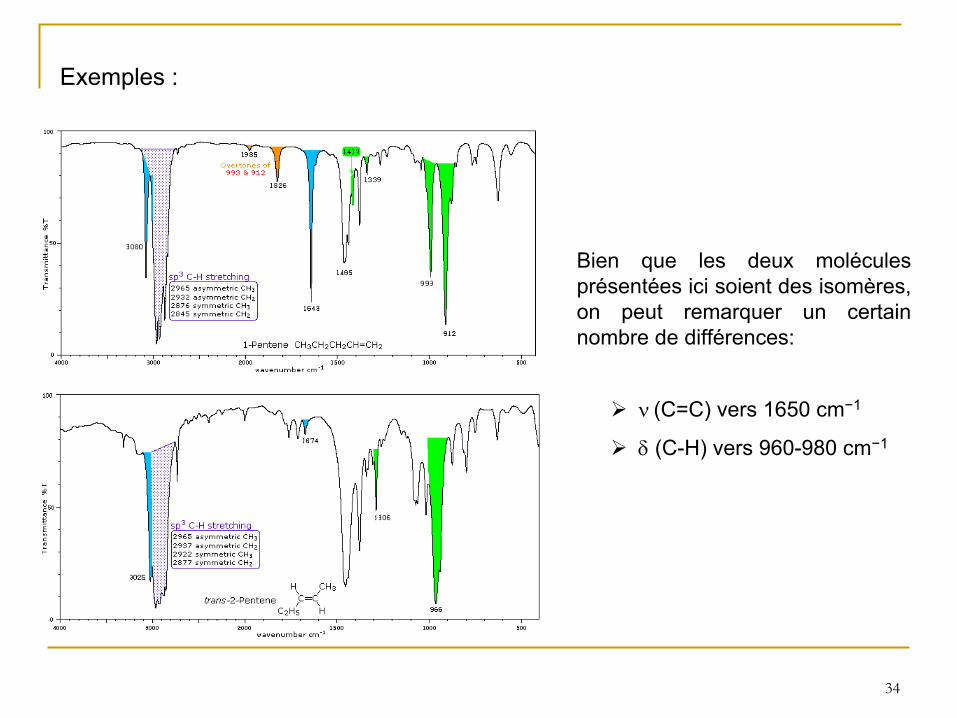
34
Exemples :
Bien que les deux molécules présentées ici soient des isomères, on peut remarquer un certain nombre de différences:
(C=C) vers 1650 cm−1
(C-H) vers 960-980 cm−1
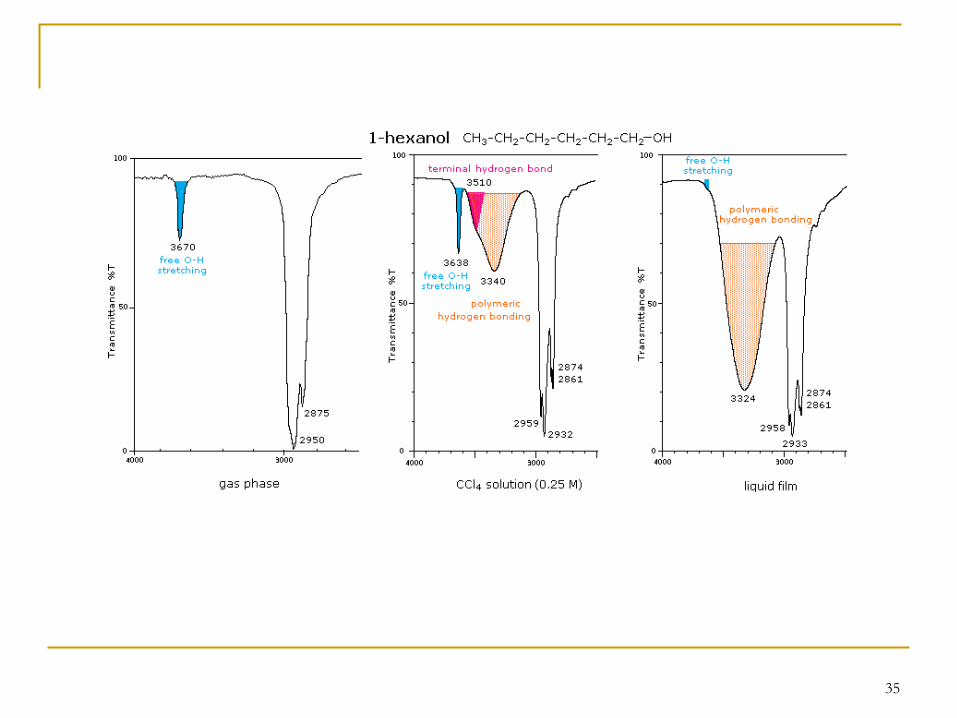
35
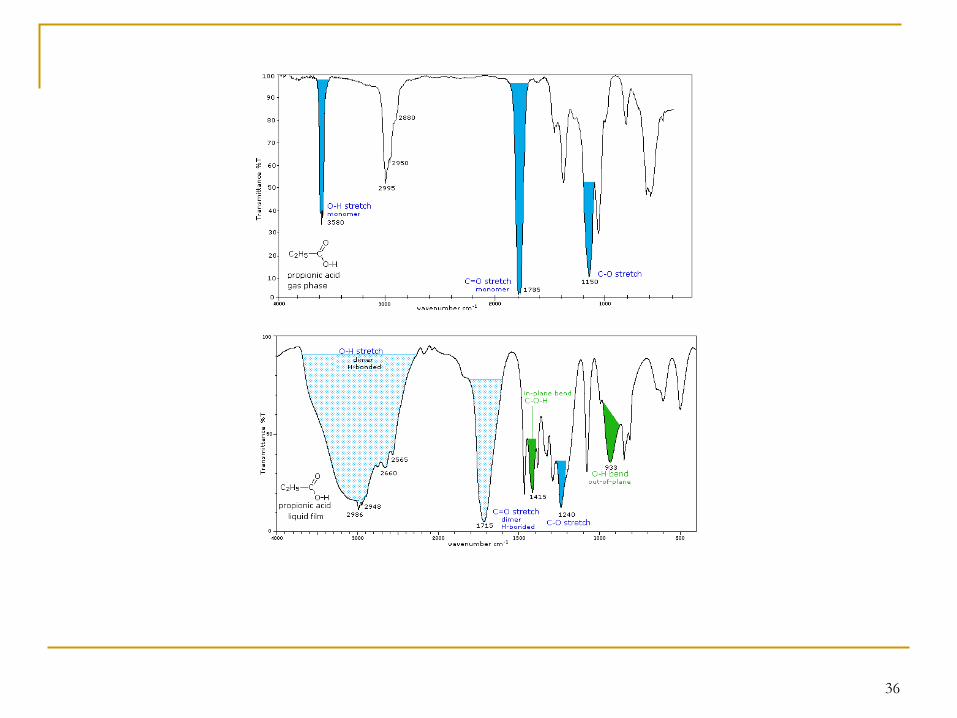
36

37

38
B/
Applications en catalyse

39
Dans cette dernière partie, nous allons discuter d’une application très importante de la spectroscopie d’absorption infrarouge : la caractérisation, par adsorption de molécules-
sondes, des catalyseurs solides.
Nous nous intéresserons ici à
deux familles de catalyseurs :
•
Les catalyseurs métalliques supportés, constitués d’un métal (noble) dispersés à
la surface d’un support (alumine, silice, etc.) ;
• Les catalyseurs acides de type zéolithes.
Pour ces analyses, la cellule infrarouge doit pouvoir être chauffée, permettant un traitement thermique des échantillons, mais elle doit aussi pouvoir être mise sous vide.
De nombreux types de cellules ont été
développés par les différents laboratoires travaillant sur le sujet.

40

41
1. Caractérisation des catalyseurs métalliques supportés
(Pt, Pd) par adsorption de CO suivie par IR

42
Les métaux supportés, tout comme les centres acides de Lewis, ne peuvent être étudiés directement en spectroscopie IR. On utilise donc des molécules-
sondes, dont l’interaction avec le métal nous informera sur les propriétés de celui-ci. Le monoxyde carbone CO est de loin la molécule la plus utilisée pour ce type d’étude, du fait de la diversité
des renseignements obtenus :
• Quantité
d’atomes de surface (dispersion)
• Etat d’oxydation du métal en surface
• Alliage
• ...
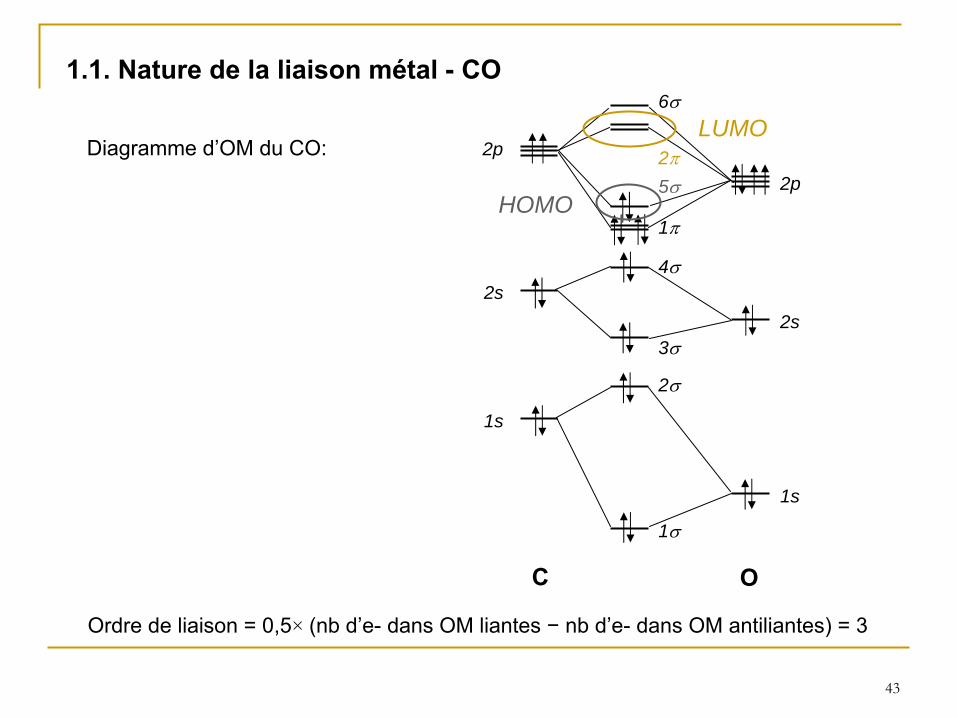
43
1.1. Nature de la liaison métal -
CO
2p
2p
2s2s
1s
1s
C O
1
2
3
4
5
6
2
1
LUMO
HOMO
Diagramme d’OM du CO:
Ordre de liaison = 0,5×
(nb d’e-
dans OM liantes −
nb d’e-
dans OM antiliantes) = 3

44
Le monoxyde de carbone forme des complexes avec presque tous les
éléments de transition. Il est généralement admis que la liaison dans ces complexes carbonyles implique la formation d’une liaison
et d’une liaison
(rétrodonative).
La liaison
est formée par le recouvrement de la plus haute orbitale pleine (HOMO) de
CO et d’une orbitale vide du métal, tandis que la liaison
est obtenue par recouvrement d’une orbitale pleine du métal de symétrie appropriée avec la plus basse orbitale vacante (LUMO) de CO (2 , antiliante).
Cette rétrodonation a pour effet de diminuer l’ordre de la liaison C−O, donc la fréquence de vibration CO . Cette fréquence passe ainsi de 2143 cm−1
pour la molécule libre (en phase gaz) à
1800 -
2100 cm−1
pour les complexes carbonyles.
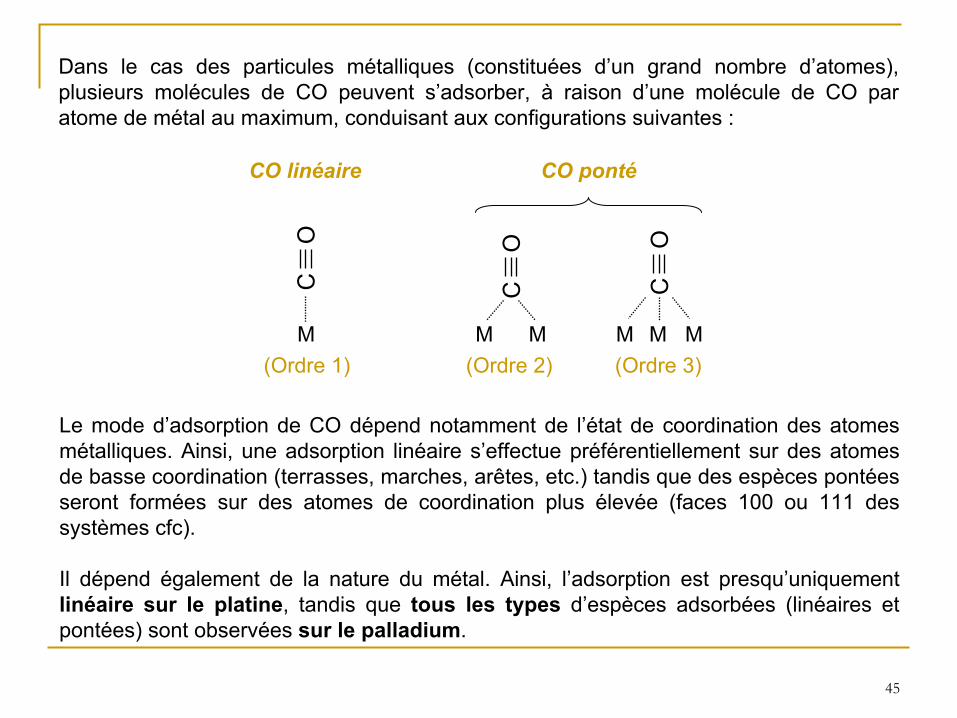
45
Dans le cas des particules métalliques (constituées d’un grand nombre d’atomes),
plusieurs molécules de CO peuvent s’adsorber, à
raison d’une molécule de CO par atome de métal au maximum, conduisant aux configurations suivantes :
CO linéaire CO ponté
(Ordre 1) (Ordre 2) (Ordre 3)M
CO
MM
CO
M MM
CO
Le mode d’adsorption de CO dépend notamment de l’état de coordination des atomes métalliques. Ainsi, une adsorption linéaire s’effectue préférentiellement sur des atomes de basse coordination (terrasses, marches, arêtes, etc.) tandis que des espèces pontées seront formées sur des atomes de coordination plus élevée (faces 100 ou 111 des systèmes cfc).
Il dépend également de la nature du métal. Ainsi, l’adsorption est presqu’uniquement linéaire sur le platine, tandis que tous les types
d’espèces adsorbées (linéaires et pontées) sont observées sur le palladium.

46
1.2. Spectres IR 1.2.1. Mode opératoire
La chimisorption de monoxyde de carbone (CO) est effectuée à
température ambiante sur une pastille de catalyseur (2 cm2
environ) préalablement réduit sous hydrogène.
Des doses croissantes de CO sont injectées sur le catalyseur, jusqu’à
saturation, avec enregistrement d’un spectre à
chaque dose (2 minutes d’équilibrage).
Les positions et les surfaces des bandes relatives aux vibrations de la liaison CO, qui apparaissent dans le spectre après adsorption, sont ensuite déterminées sur des spectres de différence :
Spectre après adsorption −
spectre avant adsorption

47
2100 2000 1900
Nombre d’onde (cm−1)
0,01
1% Pd
/
Al2
O3
1.2.2. Modes et sites d’adsorption de CO
1% Pt / Al2
O3
0,01
1900 2000 2100
Nombre d’onde (cm−1)
CO linéaire
CO ponté
CO linéaire
CO ponté
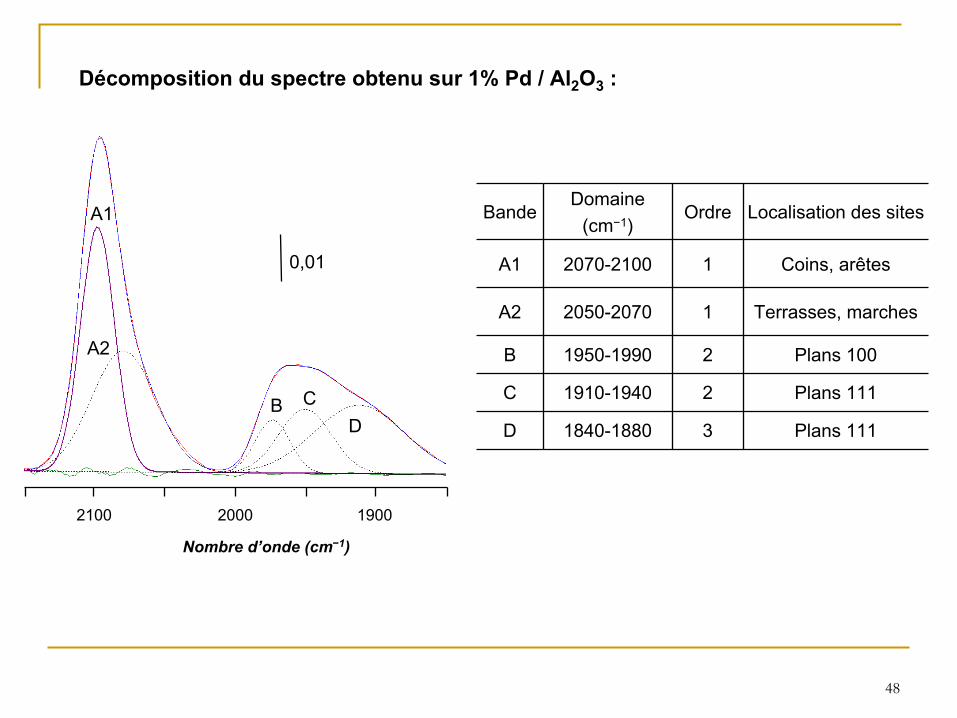
48
0,01
2100 2000 1900
A1
A2
B CD
Nombre d’onde (cm−1)
BandeDomaine
(cm−1)Ordre Localisation des sites
A1 2070-2100 1 Coins, arêtes
A2 2050-2070 1 Terrasses, marches
B 1950-1990 2 Plans 100
C 1910-1940 2 Plans 111
D 1840-1880 3 Plans 111
Décomposition du spectre obtenu sur 1% Pd / Al2
O3
:
49
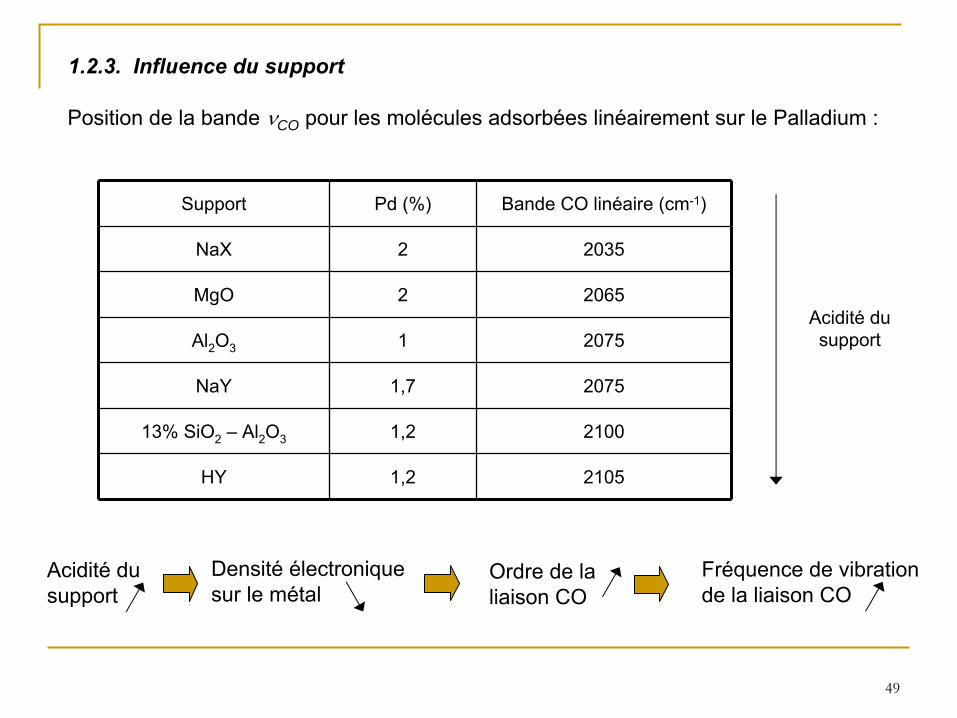
Position de la bande CO pour les molécules adsorbées linéairement sur le Palladium :
Support Pd (%) Bande CO linéaire (cm-1)
NaX 2 2035
MgO 2 2065
Al2
O3 1 2075
NaY 1,7 2075
13% SiO2
– Al2
O3 1,2 2100
HY 1,2 2105
Acidité
du support
1.2.3. Influence du support
Acidité
du support
Densité
électronique sur le métal
Ordre de la liaison CO
Fréquence de vibration de la liaison CO

50
1.2.4. Influence du taux de recouvrement
La fréquence de vibration de CO adsorbée se déplace en fonction du taux de recouvrement.
Ainsi sur le platine, la bande correspondant au CO adsorbé
linéairement passe de 2063 à
2100 cm−1
lorsque le taux de recouvrement augmente.
Une évolution contraire se produit sur l’argent, le cuivre, l’or, etc.

51
1.3. Mesure de la dispersion1.3.1. Définition
Par définition, la dispersion d’une espèce métallique à
la surface d’un support est le rapport :
100atomesd'totalNb
saccessibleatomes d' NbD
Le nombre total d’atomes métalliques Nt est généralement connu et/ou facilement calculable, connaissant la teneur en métal x du catalyseur:
Mx106,022
M100Nx
N 21At
avec x
en %mass et M
en g/mol
Le nombre d’atomes métalliques accessibles Nacc doit être déterminé. L’adsorption, suivie par IR, de doses croissantes de CO jusqu’à
saturation de la surface, permet d’estimer cette quantité
(en supposant une adsorption linéaire de CO).

52
0,02
0,04
0,06
0,08
0,10
0,12
Abs
orba
nce
1800 1900 2000 2100
Nombre d'onde (cm−1)
pCO
0
1
2
3
4
5
0 100 200 300 400 500
Pression cumulée (mbar)
Surf
ace
norm
alis
ée (c
m−1
)
S
S
= surface normalisée (à
10 mg) du massif 2150-1800 cm−1
à
saturation .
1.3.2. Principe

53
Dans le cas du platine, la quantité
d’atomes accessibles aux molécules de CO est donnée par l’équation :
0,2258SNAcc
100x106,0220,2258
195,09S100
Mx106,022
100,2258
S
100N
N%) (enD
321
18
t
Acc
(×
10
18
atomes / g de catalyseur)
La dispersion du platine à
la surface du catalyseur s’exprime donc :
xS14,35%) (enD
54
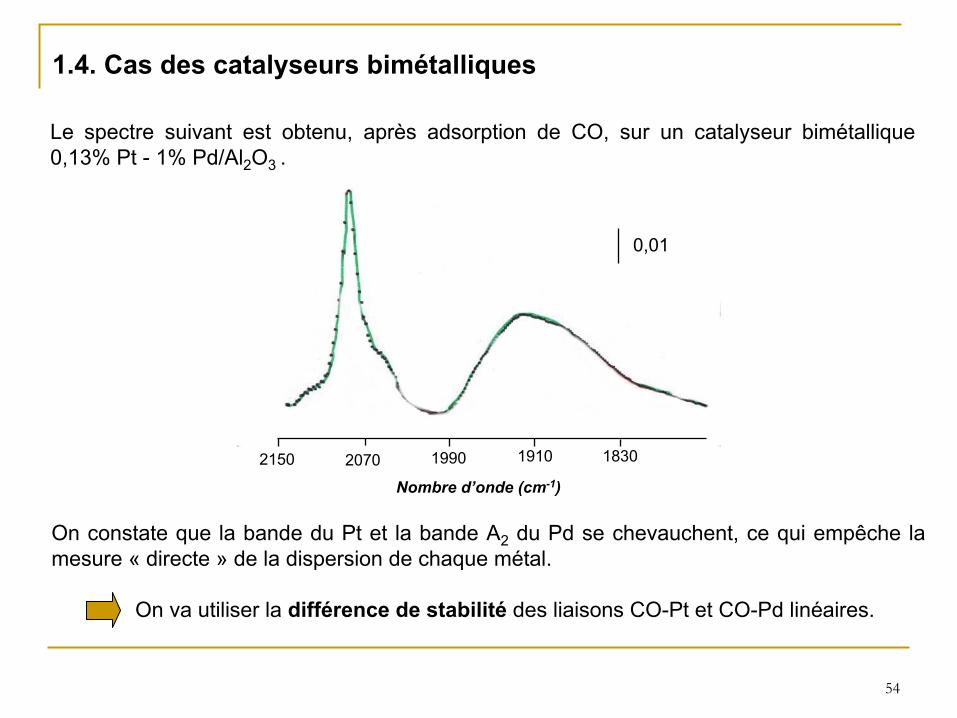
1.4. Cas des catalyseurs bimétalliques
On constate que la bande du Pt et la bande A2
du Pd se chevauchent, ce qui empêche la mesure «
directe
»
de la dispersion de chaque métal.
On va utiliser la différence de stabilité
des liaisons CO-Pt et CO-Pd linéaires.
1910 18302150 2070 1990
Nombre d’onde (cm-1)
Le spectre suivant est obtenu, après adsorption de CO, sur un catalyseur bimétallique 0,13% Pt -
1% Pd/Al2
O3 .
0,01

55
Les espèces CO adsorbées linéairement sur le platine sont fortement liées et résistent à
un dégazage sous vide, tandis que celles adsorbées linéairement sur le palladium (A1 et A2), plus faiblement liées, sont éliminées.
1910 18302150 2070 1990
Nombre d’onde (cm−1)
1750
0,020,02
2150 2050 1950
Nombre d’onde (cm−1)
0,13% Pt/Al2
O3 1% Pd/Al2
O3
La figure suivante montre l’effet d’un dégazage sous vide (10−3
torr) sur la quantité
de CO adsorbée sur les catalyseurs monométalliques :

56
L’analyse des spectres avant et après dégazage permettra, par différence, de d’estimer le nombre d’atomes de Pd et de Pt accessibles à
la surface du catalyseur bimétallique et donc d’évaluer la dispersion de chacun.
1910 18302150 2070 1990
0,01avant dégazage
après dégazage
Nombre d’onde (cm−1)

57

58
2. Caractérisation des catalyseurs acides par adsorption de
molécules-sondes suivie par IR

59
Contrairement à
la caractérisation des catalyseurs métalliques supportés qui emploie quasi exclusivement CO comme molécule-sonde, la caractérisation des propriétés acides des catalyseurs fait appel à
un grand nombre de molécules-sondes.
Cette variété
s’explique en partie par la volonté
d’adapter la sonde au catalyseur, en faisant varier la basicité
de la molécule-sonde, ainsi que sa taille.
Les molécules les plus couramment utilisées sont :
• La pyridine
• L’ammoniaque
• Le monoxyde de carbone
N
NH
H H
Comme nous le verrons par la suite, cette liste n’est pas exhaustive ...

60
1.1. Définitions d’un acide
• Brönsted et Lowry: un acide est une espèce capable de céder un proton H+.
•
Lewis: un acide est une espèce capable de recevoir un doublet d’électrons (définition plus générale).
On pourra donc distinguer deux types
de «
sites
»
acides, des sites de Brönsted (acidité
protonique) et des sites de Lewis.
Or, la plupart des réactions (industrielles) catalysées par des acides solides ne font intervenir que les sites protoniques. Il conviendra donc, très souvent, de distinguer les fonctions acides protoniques (acides de Brönsted) et les fonctions acides non protoniques (acides de Lewis).
C’est particulièrement vrai dans le cas des zéolithes, qui constituent, de loin, le catalyseur solide acide le plus utilisé
industriellement (l’autre étant l’alumine chlorée).

61
1.2. Les zéolithes
Les zéolithes sont des silico-aluminates parfaitement cristallisés, appartenant à
la famille des tectosilicates, de formule générale :
Mx
Sin−x
Alx
O2n
, y H2
O
où
M est un cation alcalin monovalent (Na+, K+).
Le rapport atomique Si/Al est toujours supérieur à
l’unité
(règle de Loewenstein) et le nombre y de molécule d’eau par maille dépend de la nature de la zéolithe, ainsi que du rapport Si/Al.
Les zéolithes sont formées d’un arrangement tridimensionnel
de tétraèdres SiO4
et AlO4
−, liés par leurs atomes d’oxygène. Il en résulte une structure poreuse très ouverte et régulière, constituée de canaux et/ou de cages généralement inférieures à
0,8 nm de diamètre, donc de taille voisine des molécules organiques.
L’élimination des molécules d’eau présentes dans la structure, par un simple chauffage (d’où
le nom de zéolithe, «
pierre qui bout
»), libère donc un volume microporeux très important, qui peut être mis à
profit dans des opérations diverses : séchage, purification, séparation et transformation catalytique.
1.2.1. Généralités
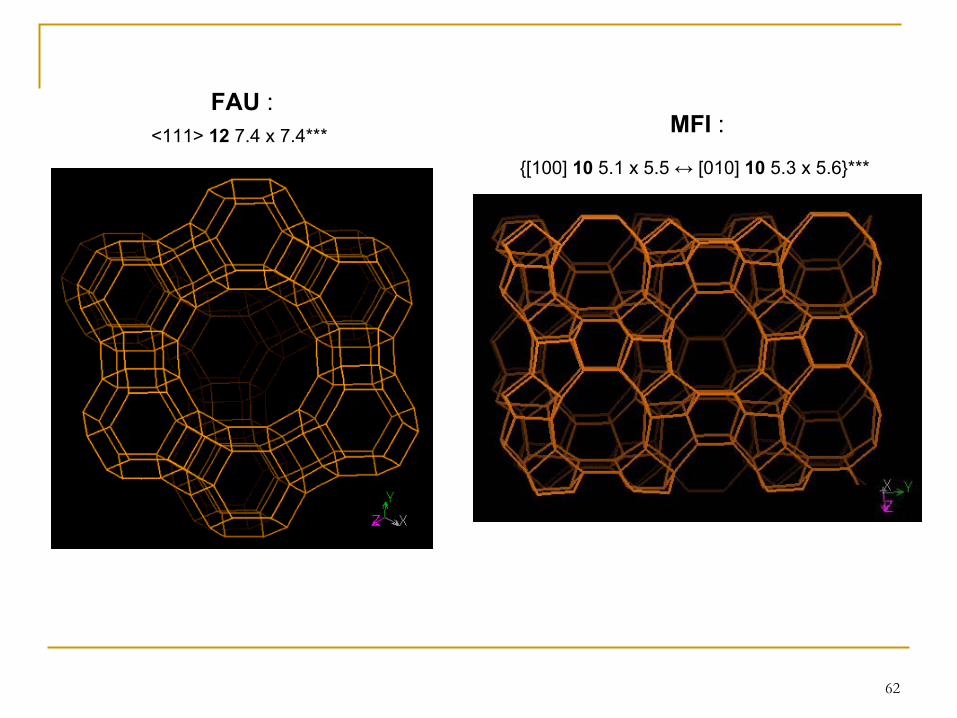
62
{[100] 10
5.1 x 5.5 ↔ [010] 10
5.3 x 5.6}***
MFI
:FAU
:
<111> 12 7.4 x 7.4***

63
A ce jour, une cinquantaine de zéolithes ont été
découvertes à
l’état naturel, mais plus de 170 structures différentes ont été
synthétisées. Parmi celles-ci, 17 sont produites industriellement.
En tonnage, la zéolithe synthétique la plus vendue est la zéolithe 4A (LTA), utilisée en remplacement des polyphosphates de sodium dans les lessives. Par
contre, le marché
le plus important en valeur concerne l’utilisation des zéolithes synthétiques comme catalyseurs.
Les zéolithes de type FAU (H-Y), utilisées en grande quantité
dans le procédé
FCC (Fluid Catalytic Cracking), qui permet la conversion des fractions pétrolières lourdes en essence, représentent 95 % du marché
des catalyseurs.
Les autres types de zéolithes très largement utilisés sont les zéolithes MFI (ZSM-5), BEA (), MOR et MWW (MCM-22).
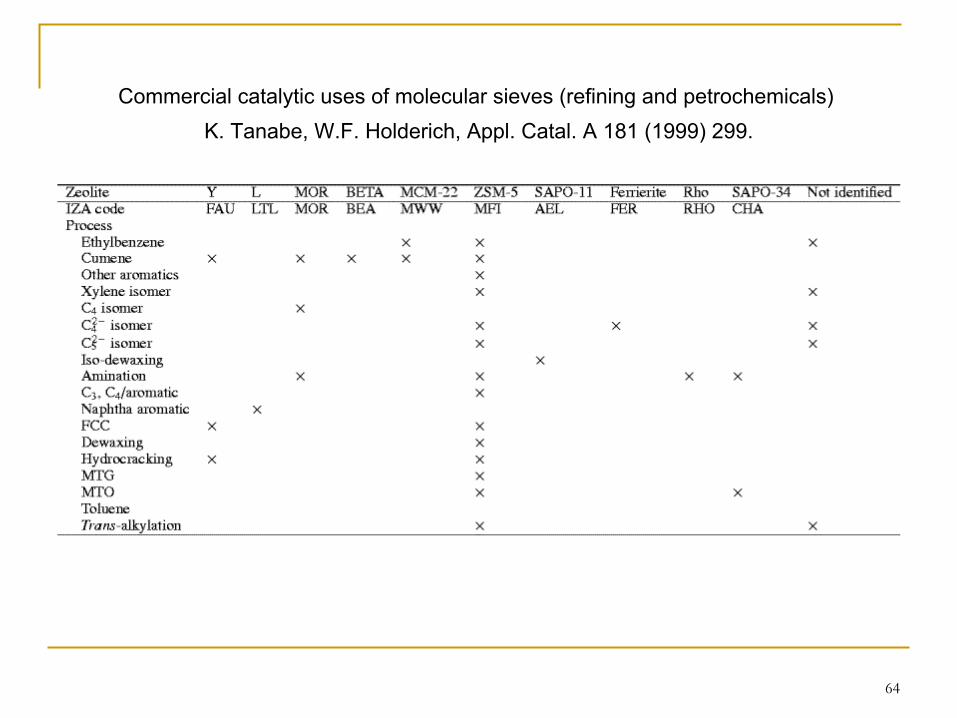
64
Commercial catalytic uses of molecular sieves (refining and petrochemicals) K. Tanabe, W.F. Holderich, Appl. Catal. A 181 (1999) 299.

65
On a souvent pour habitude de classer les zéolithes en fonction de la taille de leurs ouvertures. On distingue :
•
Les zéolithes à
petits pores, qui présentent des ouvertures de pores définies par 8 atomes d’oxygène (diamètre compris entre 0,3 et 0,45 nm)
•
Les zéolithes à
taille de pores intermédiaire, d’ouverture définie par 10 atomes d’oxygène (diamètre compris entre 0,45 et 0,6 nm)
•
Les zéolithes à
larges pores, d’ouverture définie par 12 atomes d’oxygène (diamètre compris entre 0,6 et 0,8 nm)

66
1.2.2. Acidité
des zéolithes
Du fait de la coordinance 4 de l’aluminium et de sa valence 3, le tétraèdre AlO4−
est porteur d’une charge négative, compensée par la présence d’un ion compensateur (en général Na+
ou K+). En catalyse, ces cations sont échangés par des protons H+, ce qui confère une acidité
de Brönsted à
la zéolithe.
A chaque tétraèdre aluminique correspond donc potentiellement un site acide de Brönsted.
La concentration (densité) en sites de Brönsted joue bien évidemment un rôle crucial dans l’activité
catalytique des zéolithes mais celle-ci dépend également de la force
des sites.
Z−, Na+ Z−, NH4+ Z−, H+
NH4
NO3 400 –
600°C
−
NH3

67
Les deux principaux paramètres qui régissent la force acide des sites de Brönsted sont :
•
un facteur géométrique
: une augmentation de l’angle
Si(OH)Al
diminue le caractère covalent de la liaison OH et donc renforce l’acidité
du groupement hydroxyle. La valeur de cet angle et la longueur des liaisons Si-O-Al dépendent du type de structure. Ainsi, l’angle SI-O-Al est plus élevé
dans le cas des zéolithes MOR et MFI que dans celui de la zéolithe FAU, conférant à
ces deux zéolithes une plus grande force acide de Brönsted.
•
un facteur de composition chimique
: le nombre de tétraèdres aluminiques seconds voisins joue un rôle important sur la force du site acide de Brönsted considéré. Plus ce nombre est élevé
(et donc plus le rapport Si/Al est faible) , plus l’électronégativité
de la charpente, et donc la force acide, est faible.

68
Toutefois, au cours de la préparation de la zéolithe ou de leur utilisation, une altération de la charpente zéolithique peut se produire, se traduisant par l’extraction d’une partie de l’aluminium. Les conséquences possibles sont:
• la création d’espèces aluminiques extraréseau
(AlER), qui sont des acides de Lewis
•
une diminution de la concentration en sites acides de Brönsted, avec éventuellement une augmentation de leur force acide
•
dans certains cas, une baisse de l’accessibilité
des réactifs à
la porosité, due à
la présence d’AlER.
•
une exaltation de la force acide
des sites de Brönsted
se situant à
proximité
de sites de Lewis.
• la concentration en sites de Brönsted (et de Lewis)
• la force
de ces sites
• l’accessibilité
de ces sites
La caractérisation (complète) de l’acidité
des zéolithes requiert donc d’étudier trois paramètres:

69
1.3. Caractérisation de l’acidité
des zéolithes par spectroscopie IR
1500 2000 2500 3000 3500
Nombre d'onde (cm−1)
Si-O
O-H
0,05
Exemple:
Spectre FT-IR obtenu après dégazage sur un échantillon de zéolithe MCM-22
Pour ces études, l’échantillon de zéolithe requiert un traitement thermique préalable, afin d’éliminer l’eau contenue dans les pores. Ce traitement consiste en un chauffage (450°C) sous flux d’air pendant une nuit, puis un dégazage sous vide secondaire (10-3
torr) pendant 2 h. Les spectres sont ensuite enregistrés à
T ambiante.
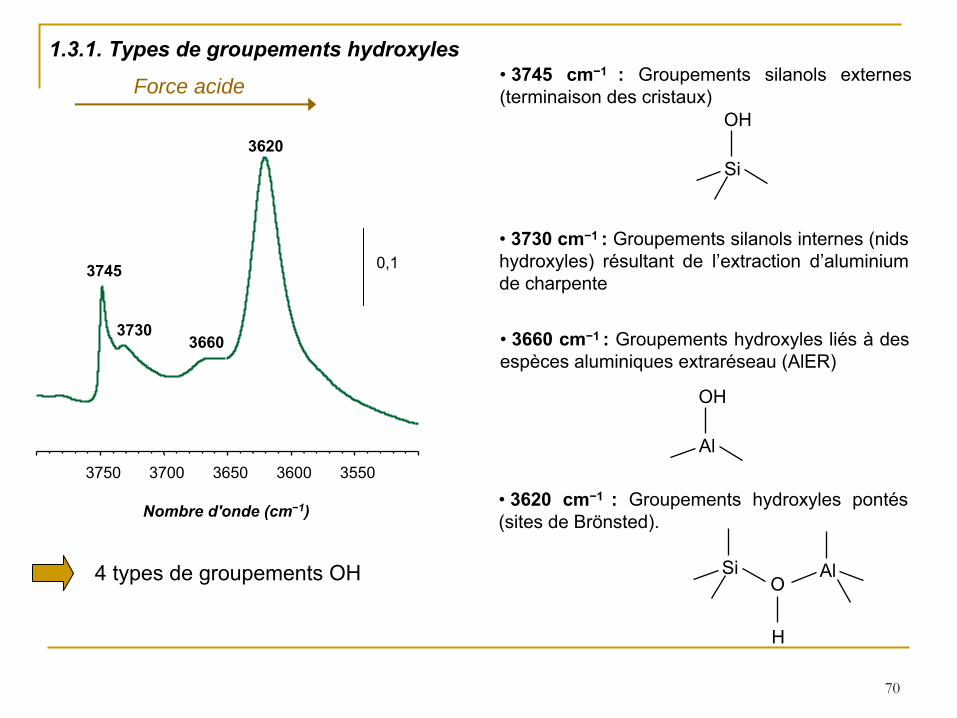
70
3550 3600 3650 3700 3750
0,1
3620
3660 3730
3745
Nombre d'onde (cm−1)
4 types de groupements OH
•
3745 cm−1 :
Groupements silanols externes (terminaison des cristaux)
Si
OH
•
3730 cm−1 :
Groupements silanols internes (nids hydroxyles) résultant de l’extraction d’aluminium de charpente
•
3660 cm−1 :
Groupements hydroxyles liés à
des espèces aluminiques extraréseau (AlER)
Al
OH
•
3620 cm−1 :
Groupements hydroxyles pontés (sites de Brönsted).
SiO
Al
H
Force acide
1.3.1. Types de groupements hydroxyles

71
1.3.2. Adsorption de pyridine
La pyridine est la molécule-sonde la plus utilisée pour la caractérisation de l’acidité
des zéolithes. Toutefois, sa taille assez importante (0,57 nm) restreint son utilisation aux zéolithes à
larges pores et à
certaines zéolithes à
taille de pores intermédiaires.
C’est une base relativement forte (pKa = 5,2) qui :
• se protone sur les sites de Brönsted (formant un ion pyridinium)
• se coordine aux sites de Lewis
• forme des liaisons hydrogène avec des groupements OH peu acides (silanols)
A ces modes d’adsorption chimique, s’ajoute une adsorption physique, que l’on doit éliminer (par traitement thermique) pour ne pas être gêné.
L’adsorption de pyridine peut s’effectuer selon deux méthodes:
• ajouts dosés, méthode précise mais assez fastidieuse
• saturation de l’échantillon
72
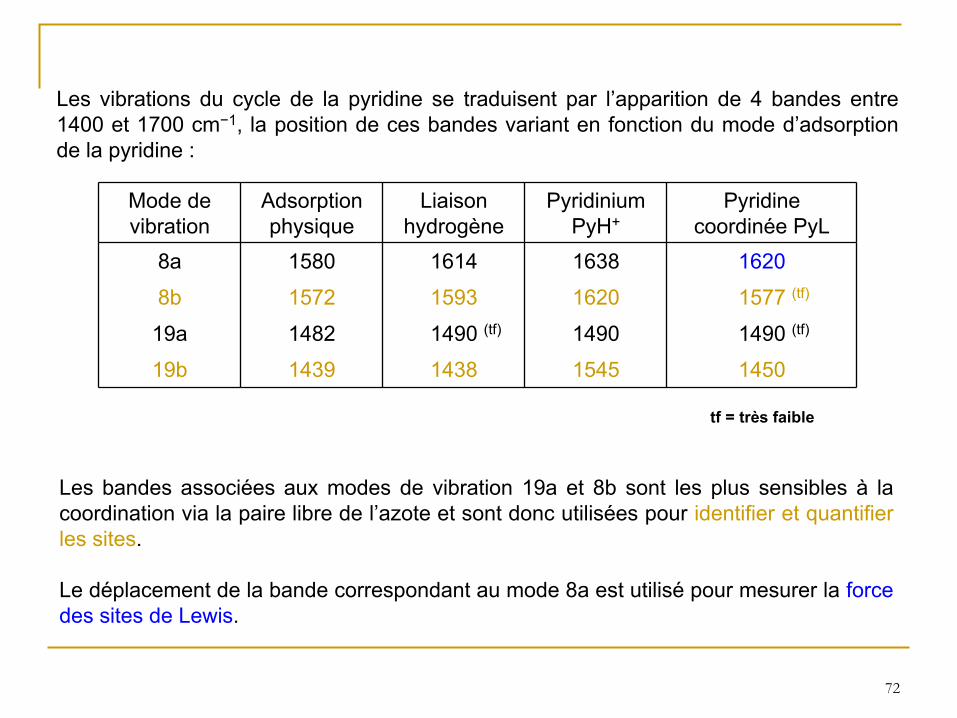
Les vibrations du cycle de la pyridine se traduisent par l’apparition de 4 bandes entre 1400 et 1700 cm−1, la position de ces bandes variant en fonction du mode d’adsorption de la pyridine :
Mode de vibration
Adsorption physique
Liaison hydrogène
Pyridinium PyH+
Pyridine coordinée PyL
8a 1580 1614 1638 1620
8b 1572 1593 1620 1577 (tf)
19a 1482 1490 (tf) 1490 1490 (tf)
19b 1439 1438 1545 1450
Les bandes associées aux modes de vibration 19a et 8b sont les plus sensibles à
la coordination via la paire libre de l’azote et sont donc utilisées pour identifier et quantifier les sites.
Le déplacement de la bande correspondant au mode 8a est utilisé
pour mesurer la force des sites de Lewis.
tf = très faible
73
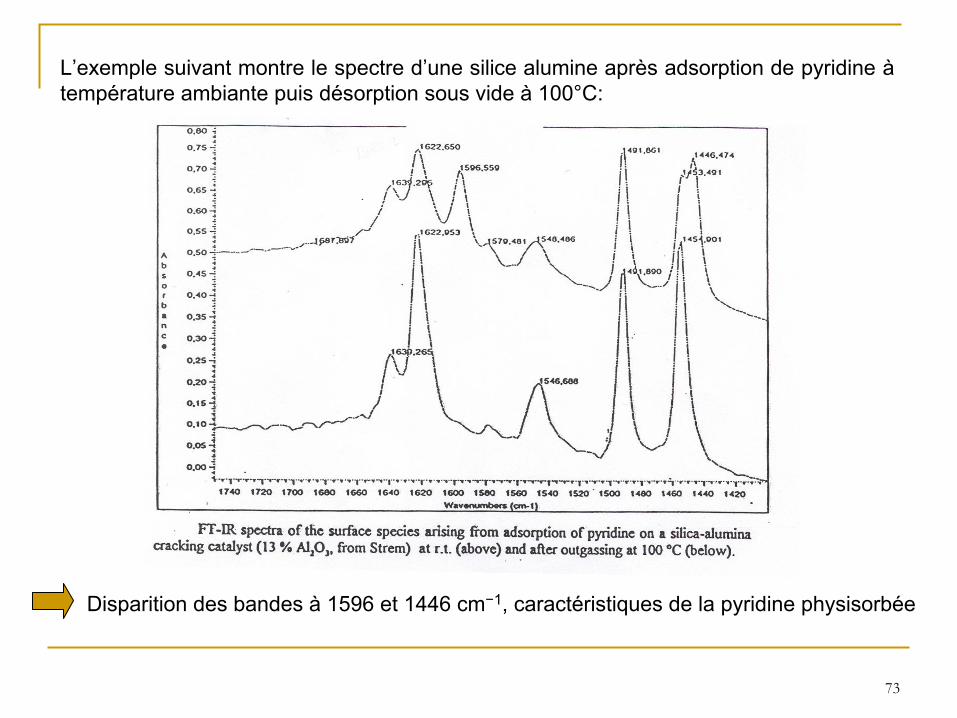
L’exemple suivant montre le spectre d’une silice alumine après adsorption de pyridine à
température ambiante puis désorption sous vide à
100°C:
Disparition des bandes à
1596 et 1446 cm−1, caractéristiques de la pyridine physisorbée
74
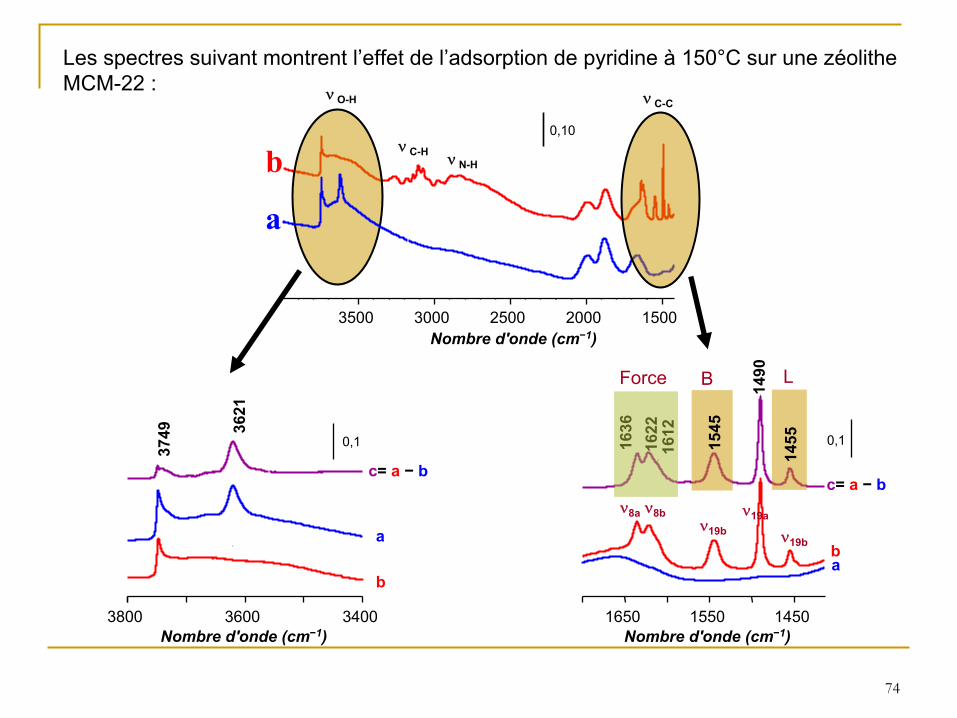
Les spectres suivant montrent l’effet de l’adsorption de pyridine à
150°C sur une zéolithe MCM-22 :
3621
3749 0,1
3400 3600 3800 Nombre d'onde (cm−1)
a
b
c= a
−
b
1490
1612
1622
1636
0,1
1450 1550 1650
1545
1455
B LForce
8a
8b19b 19b
19a
1500 2000 2500 3000 3500 Nombre d'onde (cm−1)
a
0,10
b
C-H
C-C
N-H
O-H
Nombre d'onde (cm−1)
ab
c= a
−
b

75
La détermination de la concentration Q (en µmol/g) en sites acides de Brönsted et de Lewis s’effectue à
partir des aires des bandes à
1545 et 1450 cm−1
respectivement, en utilisant la relation :
mSAQ
ε
avec : • A l’aire de la bande (en cm−1)• S la surface de la pastille (en cm2)• le coefficient d’extinction molaire (µmol/cm)• m la masse de la pastille (en g)
Le coefficient d’extinction molaire doit être déterminé
pour chaque bande, en utilisant la méthode des ajouts dosés, sur un matériau ne comportant que des sites de Brönsted (une zéolithe dépourvue de sites de Lewis) ou que des sites de Lewis (en général, une alumine).
Dans le cas de la pyridine, sa valeur est quasiment indépendante du type de matériau. Toutefois, des valeurs très différentes peuvent être trouvées dans la littérature.

76
Echantillons Références ε1545
PyH+(cm.µmol−1)
HY S. Khabtou et al, Micropor. Mater., 1994 1,8 +/-
0,1
HY Echoufi et al, Catal. Letters, 1996 0,81
HY et EMT Makarova et al, Micropor. Mater.,1995 1,1
HMOR Datka et al, J. Catal., 1992 0,73
HMOR Emeis, J. Catal., 1993 1,67 +/-
0,12
HY Thibault-Starzyk et al, Micropor. Mesopor. Mat., 20041,36
Thermogravimétrie
HMOR Guisnet et al, Polish. J. Chem., 1997 1,13

77
Enfin, une distribution en force des sites de Brönsted (voire de Lewis) peut être établie par désorption de la pyridine à
des températures croissantes :
0
0,2
0,4
0,6
0,8
1
150 250 350 450 500
1545 cm–1
(Brönsted)1450 cm–1
(Lewis)
Température de désorption de la pyridine (°C)
Surf
ace
rela
tive
de la
ban
deH-MCM-22

78
On peut en effet considérer que plus un site acide est fort, plus la liaison établie entre ce site acide et la molécule de pyridine sera forte, et donc plus la température nécessaire pour rompre la liaison sera élevée.
Dans les zéolithes, on considère que les sites capables de retenir la pyridine à
une température supérieure à
350°C sont forts. On peut remarquer que, dans l’exemple précédent, la proportion de sites de Brönsted forts est très faible (4 %), tandis qu’elle est élevée pour les sites de Lewis (61 %).
Toutefois, cette méthode est de plus en plus contestée, en particulier pour les zéolithes à
taille de pores intermédiaire. En effet, dans ce cas, la taille de la molécule de pyridine est très proche de celle des pores. Par conséquent, la désorption des molécules de pyridine est plus difficile, ce qui conduit à
une surestimation
de la quantité
de sites forts.
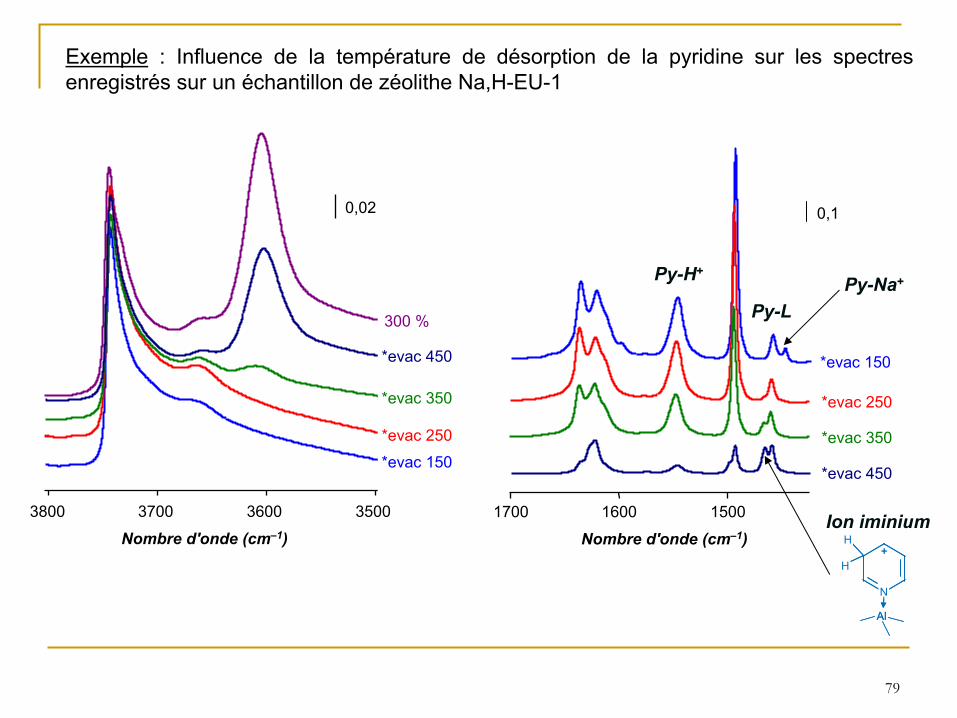
79
Exemple
: Influence de la température de désorption de la pyridine sur les spectres enregistrés sur un échantillon de zéolithe Na,H-EU-1
3500 3600 3700 3800
Nombre d'onde (cm–1)
*evac 450
*evac 350
*evac 250
*evac 150
300 %
0,02
1500 1600 1700
*evac 450
*evac 350
*evac 250
Nombre d'onde (cm–1)
*evac 150
0,1
Py-Na+
Ion iminium
N
+H
H
Al
N
+H
H
Al
Py-H+
Py-L

80
Adsorption de pyridine sur catalyseurs «
cokés
»
Choix délicat des conditions de prétraitement, en particulier pour éviter l’élimination du coke «
léger », formé
lors de réactions à
des températures «
modérées
»
(< 450°C).
Problèmes de résolution des spectres: Si %C > 3%, augmenter le nombre de scans et surveiller le niveau d’énergie du faisceau.
Au cours des réactions chimiques, les catalyseurs zéolithiques subissent une désactivation, plus ou moins rapide et importante. Cette désactivation est généralement due à
la formation de molécules polyaromatiques volumineuses («
coke
»), qui restent piégées dans les pores de la zéolithe. Il est souvent utile d’évaluer l’acidité
(et la porosité) résiduelle de ces zéolithes «
cokées
», mais aussi de déterminer la nature chimique du «
coke
».
La spectroscopie IR fournit des réponses à
ces différents problèmes. Toutefois des difficultés supplémentaires apparaissent dans le traitement de ces catalyseurs «
cokés
». Les premières sont d’ordre opératoire:
D’autres problèmes, plus délicats, apparaissent lorsque l’on veut mesurer l’acidité
par adsorption de pyridine, comme montré
sur l’exemple suivant :

81
Adsorption de pyridine sur une zéolithe H-FAU, cokée par le méthylcyclohexane :
0,1
1450 1550 1650
B L
c
c
= b -
aa : cata coké b : cata coké
+ Pyr
b
a
1350
Nombre d'onde (cm−1)
C–HC–C
•
Le spectre a indique que le «
coke
»
est constitué
d’espèces polyaromatiques (bande de «
coke
»
à
1600 cm–1) alkylées (bande à
1375 cm–1)
•
L’apparition de bandes «
négatives
»
dans le spectre c, pour des fréquences correspondant aux bandes présentes dans le spectre a, suggère la désorption d’une partie du coke lors de l’addition de pyridine.
Ce déplacement dépend de la nature de la zéolithe, ainsi que de la nature du «
coke
». Il complique bien évidemment la mesure de l’acidité
…

82
En résumé, concernant l’utilisation de la pyridine :
Différenciation des sites Brönsted et Lewis (hétérogénéité)
Reproductibilité
Quantification possible (Brönsted de Lewis et des OH)
Accessibilité
restreinte aux zéolithes à
larges pores et à
certaines de taille intermédiaire (MFI)
Peu sensible à
la force des sites
Transformation de la sonde : formation d’ion dihydropyridinium
Attention sur catalyseurs cokés
Les avantages :
Les inconvénients :
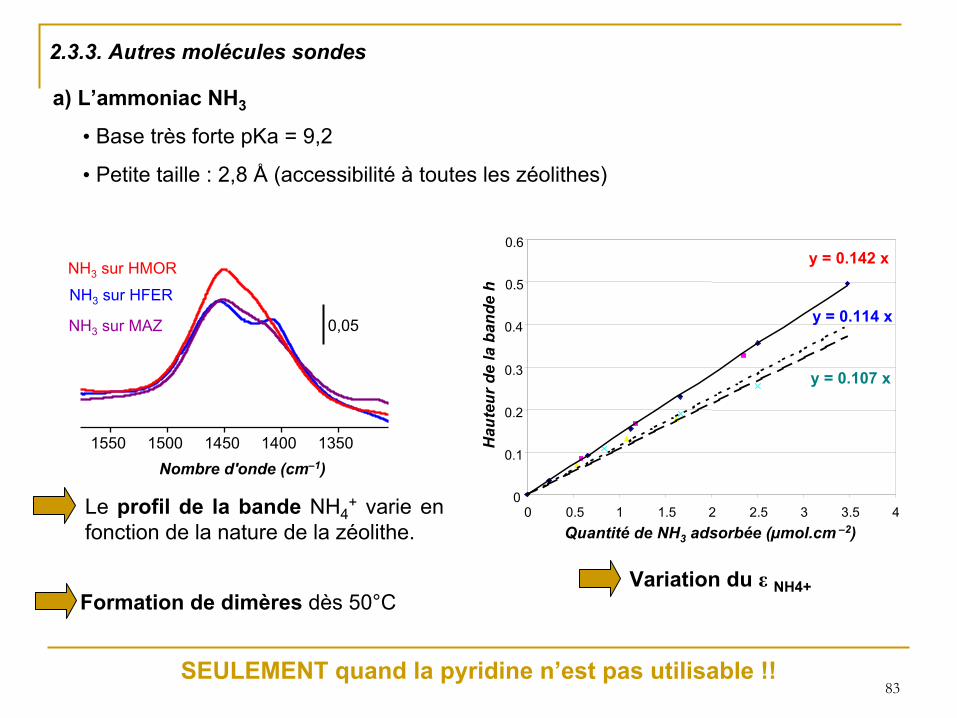
83
2.3.3. Autres molécules sondes
a) L’ammoniac NH3
• Base très forte pKa = 9,2
• Petite taille : 2,8 Å
(accessibilité
à
toutes les zéolithes)
y = 0.142 x
y = 0.114 x
y = 0.107 x
0
0.1
0.2
0.3
0.4
0.5
0.6
0 0.5 1 1.5 2 2.5 3 3.5 4Quantité
de NH3
adsorbée (µmol.cm
–2)
Hau
teur
de
la b
ande
hLe profil de la bande
NH4+
varie en fonction de la nature de la zéolithe.
NH3
sur HFER
NH3
sur HMOR
NH3
sur MAZ 0,05
1350 1400 1450 1500 1550 Nombre d'onde (cm–1)
Formation de dimères dès 50°CVariation du ε
NH4+
SEULEMENT quand la pyridine n’est pas utilisable !!

84
b) CO (Molécule sonde idéale ?)
• Base très faible et de taille réduite
• Adsorption à
basse T°C (100 K)
• Polarisation du CO vers le site acide et renforcement de la liaison CO
Déplacement de la bande CO vers des nombres d’onde plus élevés. Son importance dépend de la force des sites.
-
CO
phase gaz 2143 cm–1
- CO
physisorbé
2138 cm–1
- Sites de Brönsted: CO
de 30 à
45 cm–1
- Silanols: CO
de 15 cm–1
- Sites de Lewis: CO
jusqu’à
100 cm–1
•
De plus, la quantification des sites est possible, via la détermination de coefficients d’extinction. Exemple : ε
υ(CO)
= 2.7 cm.µmol–1 pour les sites de Brönsted)
Les seuls inconvénients sont l’obtention de spectres complexes (voir diapo suivante) et les conditions opératoires requises (travail à
très basse température).
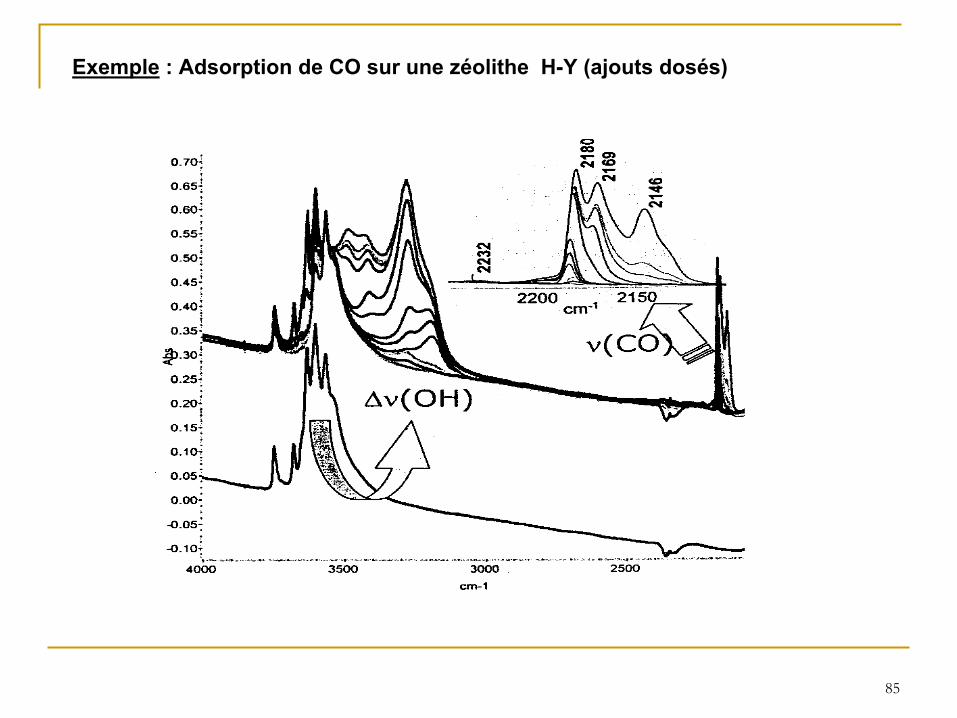
85
Exemple
: Adsorption de CO sur une zéolithe H-Y (ajouts dosés)
86
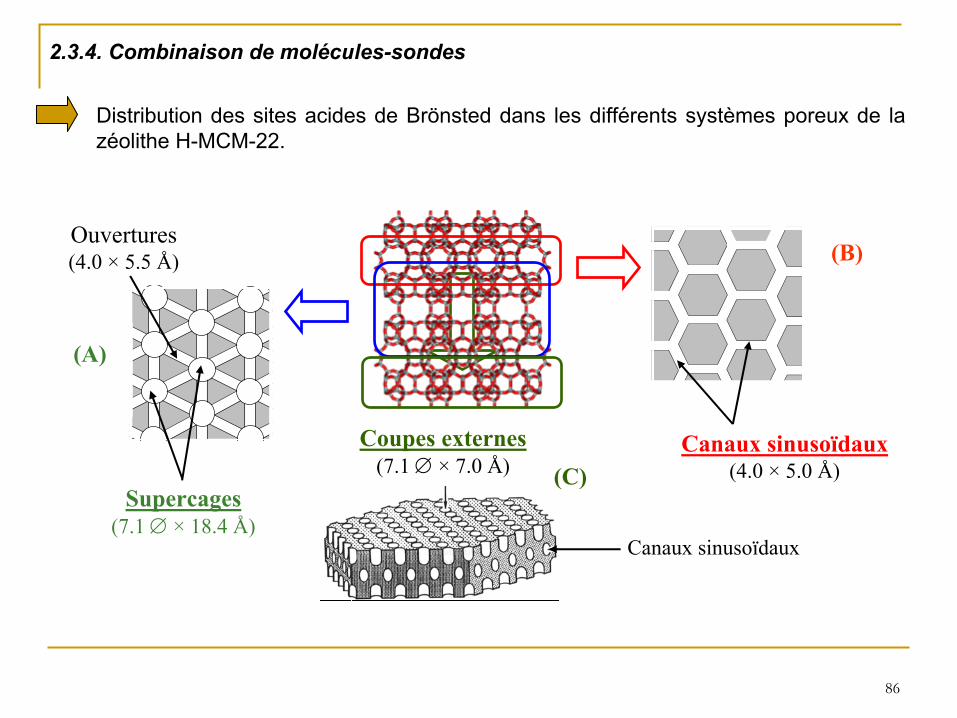
2.3.4. Combinaison de molécules-sondes
Supercages(7.1 ×
18.4 Å)
Canaux
sinusoïdaux(4.0 ×
5.0 Å)
(A)
(B)
Coupes externes(7.1 ×
7.0 Å)
Canaux
sinusoïdaux
(C)
Ouvertures(4.0 ×
5.5 Å)
Distribution des sites acides de Brönsted
dans les différents systèmes poreux de la zéolithe H-MCM-22.

87
a) Mesure de l’acidité
totale par adsorption de pyridine
Ne peut pas franchir les ouvertures des zéolithes à
taille de pores intermédiaire, telles que les
zéolithes H-MCM-22, H-ZSM-5, etc.
c) Mesure de l’acidité
externe par adsorption d’une molécule-sonde très
volumineuse: la 2,4-diméthyl-quinoléine (2,4-DMQ)
7,7 x 9,7 Å
N
CH3
CH3
3500 3600 3700
Frais
2,4-DMQ 200°C
2,4-DMQ 120°C
3500 3600 3700
Frais
2,4-DMQ 200°C
2,4-DMQ 120°C
3500 3600 3700 3500 3600 3700
Frais
2,4-DMQ 200°C
2,4-DMQ 120°C
Nombre d’onde (cm-1)
9 % des OH pontés
2,4 DMQ
1647
Abso
rban
ce
1400 1500 1600 1700 1300
0.1
Nombre d’onde (cm-1)
= 3,3 cm.µmol-1
b) Estimation de la quantité
de sites acides de Brönsted localisés dans les supercages
par adsorption de pyridine sur la zéolithe cokée (m-xylène, 24 h)
88
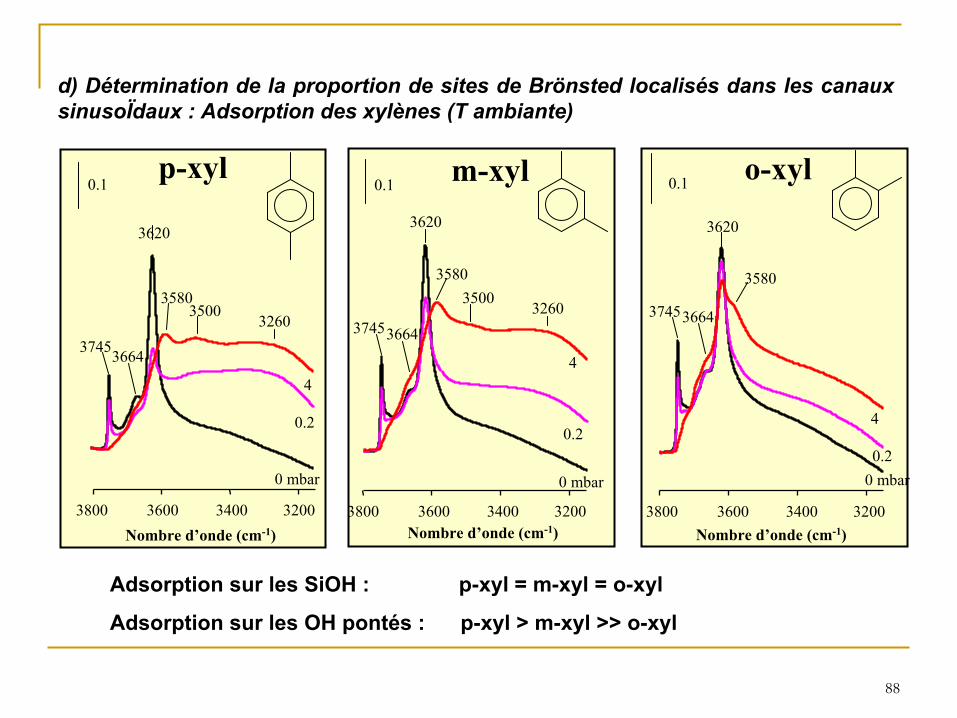
d) Détermination de la proportion de sites de Brönsted localisés dans les canaux sinusoÏdaux : Adsorption des xylènes (T ambiante)
0.1
3620
3745 3664
3580 3500 3260
0 mbar
0.2
4
3200 3400 3600 3800
m-xyl
Adsorption sur les SiOH : p-xyl = m-xyl = o-xyl
Adsorption sur les OH pontés : p-xyl > m-xyl >> o-xyl
0 mbar
3745 3664
3580 3500 3260
3620
0.1
0.2
4
3200 3400 3600 3800
p-xyl
Nombre d’onde (cm-1) Nombre d’onde (cm-1)
3745 3664
3580
3620
0.1
0 mbar0.2
4
3200 3400 3600 3800
o-xyl
Nombre d’onde (cm-1)

89
Bridging OH groups
0
0.2
0.4
0.6
0.8
1
0 1 2 3 4 8Adsorbate pressure (mbar)
Res
idua
l hei
ght o
f the
ban
d
m-Xylene p-Xylene o-Xylene
BSilanol groups
0
0.2
0.4
0.6
0.8
1
0 1 2 3 4Adsorbate pressure (mbar)
Res
idua
l hei
ght o
f the
ban
d
m-Xylene p-Xylene o-Xylene
A
Fig. 5: Adsorption of the three xylene isomers followed by IR spectroscopy. Effect of the xylene pressure on the height of the bands at 3745 cm-1
(silanols groups) and 3620 cm-1
(Bridging OH groups).
90
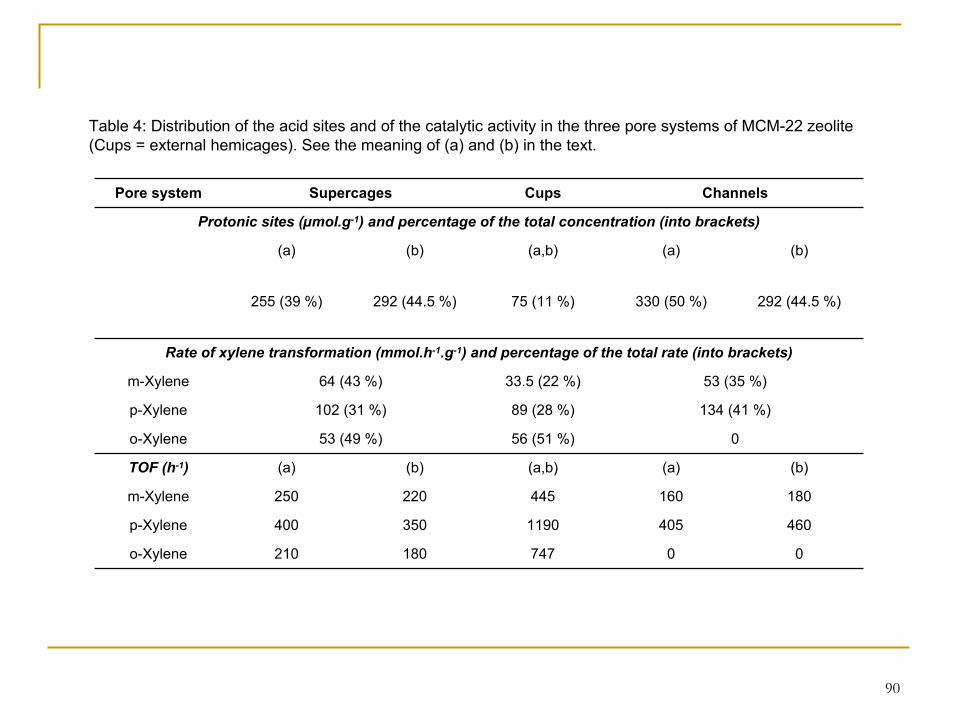
Table 4: Distribution of the acid sites and of the catalytic activity in the three pore systems of MCM-22 zeolite (Cups = external hemicages). See the meaning of (a) and (b) in the text.
Pore system Supercages Cups Channels
Protonic sites (µmol.g-1) and percentage of the total concentration (into brackets)
(a) (b) (a,b) (a) (b)
255 (39 %) 292 (44.5 %) 75 (11 %) 330 (50 %) 292 (44.5 %)
Rate of xylene transformation (mmol.h-1.g-1) and percentage of the total rate (into brackets)
m-Xylene 64 (43 %) 33.5 (22 %) 53 (35 %)
p-Xylene 102 (31 %) 89 (28 %) 134 (41 %)
o-Xylene 53 (49 %) 56 (51 %) 0
TOF (h-1) (a) (b) (a,b) (a) (b)
m-Xylene 250 220 445 160 180
p-Xylene 400 350 1190 405 460
o-Xylene 210 180 747 0 0

91
CONCLUSIONS
La variété
des molécules sondes à
notre disposition permet de bien
caractériser qualitativement et quantitativement l’acidité
des catalyseurs solides.
Localisation des sites (externes).
Être rigoureux lors du traitement des spectres.
Être prudent dans l’interprétation, surtout vis à
vis des résultats quantitatifs.
Avant toute nouvelle étude bien connaître la littérature.

92

93


















