Ecole Doctorale de Physique DEA Physique des...
153
Ecole Doctorale de Physique DEA Physique des matériaux: des nanostructures aux Grands Instruments Nanostructures Joël Cibert Laboratoire Louis Néel et Equipe "Nanophysique et semiconducteurs" septembre 2004
Transcript of Ecole Doctorale de Physique DEA Physique des...

Ecole Doctorale de Physique DEA Physique des matériaux:
des nanostructures aux Grands Instruments
Nanostructures
Joël Cibert Laboratoire Louis Néel et Equipe "Nanophysique et semiconducteurs" septembre 2004


TABLE Introduction 1 1. Les possibilités technologiques: la microélectronique; Intégration et loi de Moore 2 Les limites de l'approche technologique:
effets parasites liés aux faibles dimensions en technologie CMOS 3. Nouveaux effets grâce aux faibles dimensions
a. Approximation de la masse effective b. Effets de confinement c. Densité d'états d. Transport électrique
4. Réalisation de systèmes confinés
Première partie: élaboration I. Morphologie d'une surface cristalline 13 1. Surfaces singulières
Exemples de structures cristallines; Surfaces singulières: 2. Défauts sur une surface singulière 3. Surface vicinale 4. Reconstruction des surfaces de semiconducteurs 5 Surfaces des métaux 6. Equilibre ou cinétique? II. Surfaces modèles et modèles de croissance 19 1. Le modèle SOS (Solid on Solid)
a. Le modèle b. Surface d'équilibre à basse température c. Equations d'évolution
2. Modèle continu a. Surface rugueuse à haute température b. Modèle intermédiaire: équations d'évolution des adatomes sur une terrasse
3. Exemples d'applications du modèle SOS a. Choix de paramètres réalistes b. Résultats
4. Adatomes sur une terrasse: le modèle BCF III. Formes d'équilibre 27 1. Equilibre solide-vapeur 2. Rôle de la surface: cas d'un liquide
a. Goutte isolée (Gibbs-Thomson) b. Goutte sur un substrat plan: relation de Young:
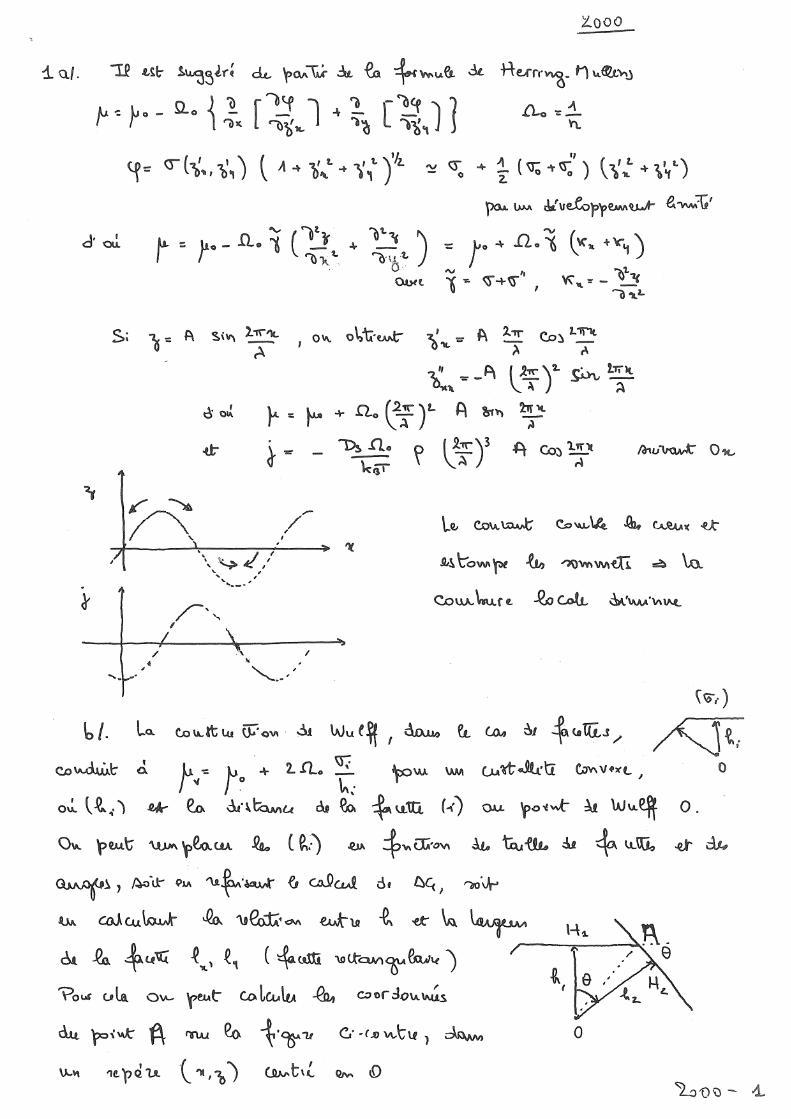
3. Solide: rôle de l'anisotropie a. Cas extrême: facettes; construction de Wulff, b. Modèle continu: potentiel chimique local c. Construction de Wulff (modèle continu) d. Différents types de croissance épitaxiale: Frank-van-der-Merwe, Volmer-Weber
4. Cinétique: oscillations RHEED, construction de Wulff dynamique IV. Hétérostructures en désaccord de maille: Contraintes et déformations élastiques 37 1. Module d'Young et coefficient de Poisson
Pression uniaxiale; Cisaillement; Pression hydrostatique

2. Notation tensorielle Le tenseur de déformations; Le tenseur de contraintes; La loi de Hooke
3. Les composantes de déformation et de contrainte 4. Quelques exemples en symétrie cubique
Pression hydrostatique; Contrainte uniaxiale; Cisaillement; Déformation dans une couche épitaxiée 5. La prise en compte des symétries 6. Modèles microscopiques. V. Relaxation des contraintes: dislocations et îlots 49 1. Dislocations 2. Dislocations de misfit et épaisseur critique 3. Ilots tridimensionnels; croissance Stranski-Krastanov
a. Déformation dans un îlot b Croissance Stranski-Krastanov c. Croissance auto-organisée
Seconde partie: propriétés électroniques I. Confinement 59 1. Systèmes 2D : confinement dans une direction
a. Puits infini : rappels b. Effet de la hauteur finie des barrières c. Réalisations
2. Boîte quantique : Confinement dans les trois directions a. Non-séparabilité b. Cas de barrières infiniment hautes c. Système à symétrie sphérique d. Un cas simple: le potentiel harmonique 3D
3. Fil quantique : confinement dans deux directions 4. Réalisations et études de boîtes quantiques
a. Boîtes autoorganisées b. Nanocristallites dans des verres ou des colloïdes
5. Interactions entre électrons a. Deux électrons dans une boîte quantique b. Atome artificiel
II. Transport 85 1. Transport diffusif et conductivité
a. Rappel : équation de Boltzmann b. Les différents types de collisions c. Gaz 2D à forte mobilité
2. Systèmes 1D ; quantification de la conductance 3. Effet tunnel
a. Fonctions d'onde en présence d'une barrière tunnel rectangulaire b. Transparence de la barrière c. Barrière de forme quelconque d. Calcul du courant. e. Calcul de perturbation et densité d'états
4. Exemples en électronique moléculaire 5. Blocage de Coulomb Bibliographie 111 Problèmes 113 Solutions exercices et problèmes 133
1
Introduction
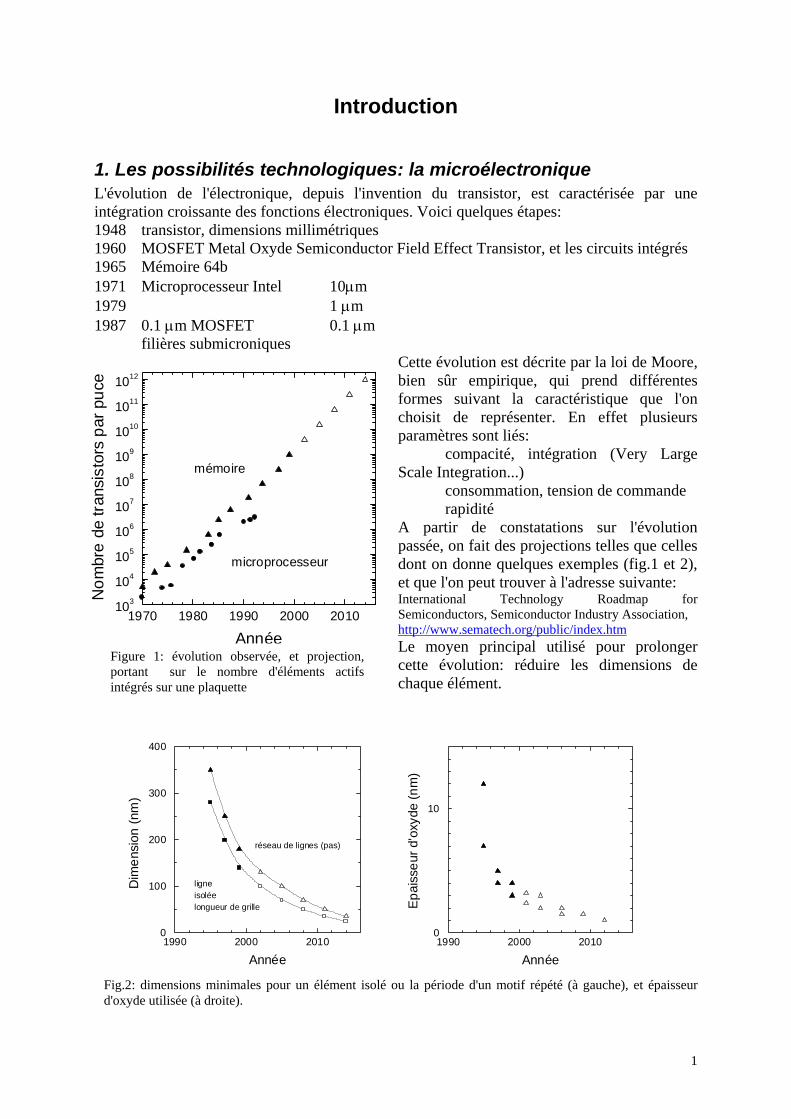
1. Les possibilités technologiques: la microélectronique L'évolution de l'électronique, depuis l'invention du transistor, est caractérisée par une intégration croissante des fonctions électroniques. Voici quelques étapes: 1948 transistor, dimensions millimétriques 1960 MOSFET Metal Oxyde Semiconductor Field Effect Transistor, et les circuits intégrés 1965 Mémoire 64b 1971 Microprocesseur Intel 10µm 1979 1 µm 1987 0.1 µm MOSFET 0.1 µm filières submicroniques
Cette évolution est décrite par la loi de Moore, bien sûr empirique, qui prend différentes formes suivant la caractéristique que l'on choisit de représenter. En effet plusieurs paramètres sont liés: compacité, intégration (Very Large Scale Integration...) consommation, tension de commande rapidité A partir de constatations sur l'évolution passée, on fait des projections telles que celles dont on donne quelques exemples (fig.1 et 2), et que l'on peut trouver à l'adresse suivante: International Technology Roadmap for Semiconductors, Semiconductor Industry Association, http://www.sematech.org/public/index.htm Le moyen principal utilisé pour prolonger cette évolution: réduire les dimensions de chaque élément.
1990 2000 20100
100
200
300
400
réseau de lignes (pas)
ligneisoléelongueur de grille
Dim
ensi
on (n
m)
Année1990 2000 2010
0
10
Epai
sseu
r d'o
xyde
(nm
)
Année
Fig.2: dimensions minimales pour un élément isolé ou la période d'un motif répété (à gauche), et épaisseur d'oxyde utilisée (à droite).
1970 1980 1990 2000 2010103
104
105
106
107
108
109
1010
1011
1012
mémoire
microprocesseur
Nom
bre
de tr
ansi
stor
s pa
r puc
e
AnnéeFigure 1: évolution observée, et projection, portant sur le nombre d'éléments actifs intégrés sur une plaquette

2
mais la description du fonctionnement de ces composants reste classique: - l'électron est considéré comme une particule de masse m*
(les effets quantiques sont oubliés après avoir défini des paquets d'ondes) - on en manipule beaucoup à la fois (par exemple, 106 dans un composant de type CCD).
2. Les limites de l'approche technologique: effets parasites liés aux faibles dimensions en technologie CMOS La figure 3 ci-dessous regroupe les éléments typiques d'un composant CMOS.
Quelles sont les limites prévisibles pour les différents éléments? la longueur de grille: les performances actuelles continuent de croître suivant les techniques utilisées: lithographie utilisant les rayons X, l'UV profond, les électrons; on développe aussi des techniques à bas coût comme le "nanoimprint"... l'épaisseur de l'oxyde de grille: On touche les limites physiques liée à l'extension des fonctions d'ondes dans la barrière d'oxyde: trois épaisseurs atomiques. les profils de dopage Les problèmes principaux sont liés à la diffusion des dopants, qui est un mécanisme statistique, et introduit donc du désordre. fluctuations On commence à ressentir les effets des fluctuations - des dimensions,
- du nombre d'impuretés de dopage, - du nombre d'électrons.
Ces limites à la technologie actuelle suscitent deux principaux axes de recherche: - améliorer la technologie et repousser ses limites (c'est ce qui a été fait jusqu'à maintenant...avec succès!) - envisager d'autres mécanismes physiques.
Fig.3: schéma d'un composant C-Mos (Nanoscale CMOS, Hon-Sum Philip Wong, D.J.Frank, P.M.Solomon, C.H.J.Wann, J.J.Welser, Proceedings of the IEEE 87 (1999) 537); à gauche, un transistor MOSFET à canal de type n, avec conduction par les électrons dans la bande de conduction; à droite, de type p, avec conduction par des trous dans la bande de valence. Le matériau est du silicium cristallin, du silicium polycristallin (contacts), de l'oxyde de silicium pour les isolants de grille.

3
3. De nouveaux effets grâce aux faibles dimensions. a. Description la plus simple: l'approximation de la masse effective Les états électroniques du solide sont décrits sous forme d'états à un électron, associés chacun à une fonction de Bloch
).exp()()( ,, rkirur knkn
rrrrrr =ψ , d'énergie
( )kEn
r. La donnée de ces énergies définit les
bandes d'énergie. Les points les plus importants pour les propriétés électroniques sont le haut de la bande de valence et le bas de la bande de conduction. En utilisant un développement limité au voisinage de ces extrema, on obtient une la dépendance quadratique (en k2) de ( )kE
r, dont on va tirer parti.
Exemple d’un semiconducteur à gap direct, GaAs. Au voisinage du bas de la bande de conduction qui est situé à 0=k , à l'énergie 0E :
- on définit une masse effective par le développement limité ( ) *0 2²²
mkEkE hr
+= , c'est-à-dire
²²
²11
* kE
m ∂∂
=h
;
- on peut écrire des équations du mouvement d'un paquet d'onde, sous l'effet d'une force appliquée F
r:
Ek∇=r
h
r 1v et Fdtkd r
h
r1
= , soit Fdtdm
rr
=v* ; on tirera parti de la similarité avec les équations
du mouvement classiques Eprrr∇=v et F
dtpd rr
= , pour oublier la mécanique quantique;
- on peut aussi décrire la fonction d'onde en introduisant la "fonction enveloppe" )()()( 0,, rFrur nkn
rrrr ≈ψ
où la fonction enveloppe F est solution de l'équation différentielle
)()()()()(*2
²0 rFEErFrVrF
mrrrrh
−=+∆− ,
ce qui permet de tenir compte d'un potentiel de perturbation )(rV r (voir partie 2). On se contentera en général dans ce cours de cette description simple. Dans les nanostructures réelles il faudra tenir compte de cas plus compliqués, par exemple: - la bande de valence, qui sera décrite sous forme de trous - éventuellement, des bandes avec une dégénérescence plus élevée
Bande de valence
Bande deconduction
Fig. 4: courbes de dispersion de GaAs (Chelikovski et Cohen, Phys. Rev. B14, 556 (1976)). Les états définissant les propriétés électroniques sont au voisinage du haut de la bande de valence (Γ8 et Γ7) et au bas de la bande de conduction (Γ6).
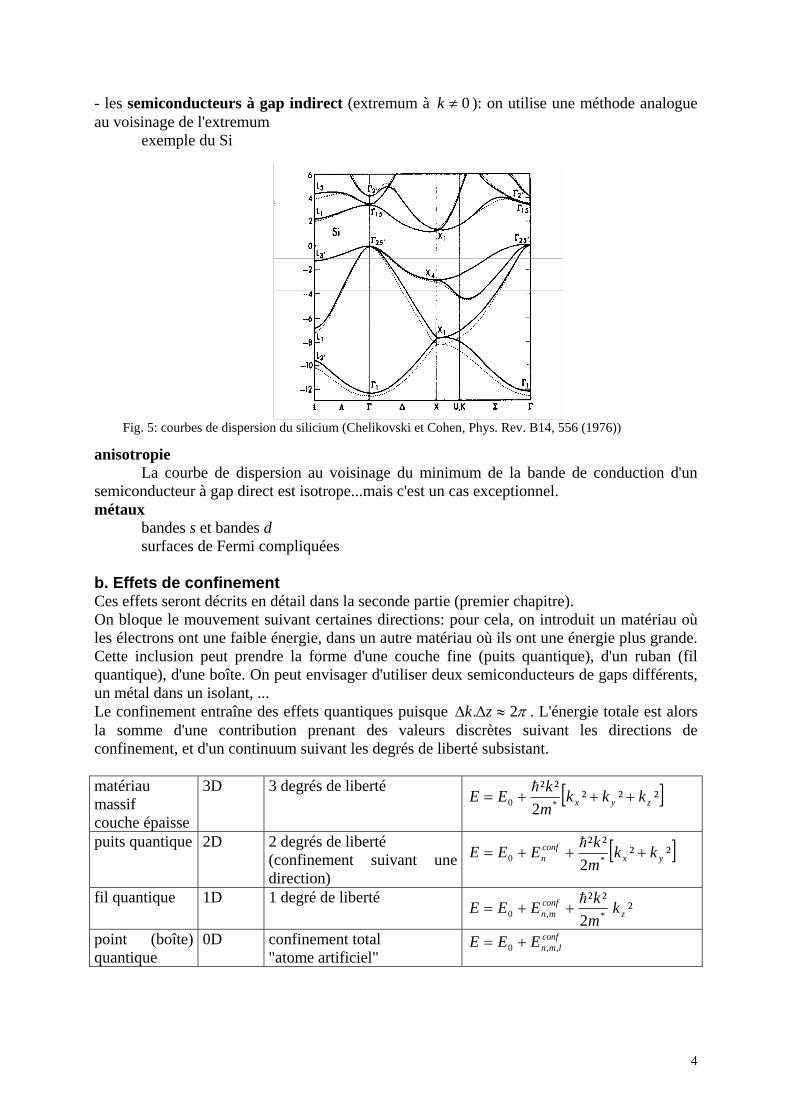
4
- les semiconducteurs à gap indirect (extremum à 0≠k ): on utilise une méthode analogue au voisinage de l'extremum exemple du Si
anisotropie La courbe de dispersion au voisinage du minimum de la bande de conduction d'un semiconducteur à gap direct est isotrope...mais c'est un cas exceptionnel. métaux bandes s et bandes d surfaces de Fermi compliquées b. Effets de confinement Ces effets seront décrits en détail dans la seconde partie (premier chapitre). On bloque le mouvement suivant certaines directions: pour cela, on introduit un matériau où les électrons ont une faible énergie, dans un autre matériau où ils ont une énergie plus grande. Cette inclusion peut prendre la forme d'une couche fine (puits quantique), d'un ruban (fil quantique), d'une boîte. On peut envisager d'utiliser deux semiconducteurs de gaps différents, un métal dans un isolant, ... Le confinement entraîne des effets quantiques puisque π2. ≈∆∆ zk . L'énergie totale est alors la somme d'une contribution prenant des valeurs discrètes suivant les directions de confinement, et d'un continuum suivant les degrés de liberté subsistant. matériau massif couche épaisse
3D 3 degrés de liberté [ ]²²²2
²²*0 zyx kkk
mkEE +++=
h
puits quantique 2D 2 degrés de liberté (confinement suivant une direction)
[ ]²²2
²²*0 yx
confn kk
mkEEE +++=
h
fil quantique 1D 1 degré de liberté ²
2²²*,0 z
confmn k
mkEEE h
++=
point (boîte) quantique
0D confinement total "atome artificiel"
conflmnEEE ,,0 +=
Fig. 5: courbes de dispersion du silicium (Chelikovski et Cohen, Phys. Rev. B14, 556 (1976))

5
c. Densité d'états La définition de la "densité d'états" est une fonction de l'énergie, notée ( )Eρ , telle que pour toute fonction f ne dépendant que de l'énergie, on a ( ) ( )dEEEff ρ∫= . Or, en considérant
les états d'une bande ( )kEr
repérés par leur vecteur d'onde kr
,avec des conditions aux limites
périodiques, et un espace de dimension d, on a aussi ( )[ ]kEfkdLf dd r∫⎟
⎠⎞
⎜⎝⎛=π2
, qu'on peut
récrire sous la forme ( ) ( )[ ]kEEEfdEkdLf dd r
−⎟⎠⎞
⎜⎝⎛= ∫∫ δπ2
. On en extrait la formule
générale de la densité d'états [ ]∑ ∫∫∫ −⎥⎦⎤
⎢⎣⎡=
nn
dd
kEEkdLE )(2
)(r
δπ
ρ (où on a rajouté l'indice
n qui désigne chacune des bandes contribuant). Dans l'hypothèse d'une masse effective isotrope, ( ) *22
0 2mkEkE hr
+= , et d’une dégénérescence gs de spin seulement, on obtient:
3D 03
2/3*
3)2(
²4)( EEmg
VE sD −=
hπρ
2D )(2
)( 02
*
2 EEYmgSE s
D −=hπ
ρ
1D 0
2/1*
11)2(
2)(
EEmg
LE sD
−=
hπρ
On remarquera que la densité d'états est nécessairement proportionnelle au "volume" de matériau envisagé (V, S ou L, suivant la dimensionalité du problème; de façon générale, Ld). Ensuite, par un simple argument d'équation aux dimensions (au sens, unité à utiliser, en l'occurrence la densité d'états est un nombre par intervalle d'énergie), on s'attend à ce que la densité d'états soit proportionnelle à
( ) EmELd
h2 et seul le facteur numérique sans dimension reste à calculer. Les formes de ces densités d'états ont des conséquences sur:
- les propriétés électroniques - les probabilités de transitions optiques (absorption, émission de lumière): laser
d. Transport électrique: Ces propriétés seront décrites dans la seconde partie (second chapitre). Le transport électrique dans un matériau s'apparente à une marche au hasard: l'électron (décrit de façon classique) subit des collisions avec des défauts, avec les phonons, avec d'autres électrons. Dans un semiconducteur, les porteurs libres sont introduits par le dopage, c'est-à-dire l'introduction d'impuretés de type donneur ou accepteur. Ceci entraîne la coexistence des porteurs libres et des impuretés chargées, avec une forte interaction. Les propriétés de transport sont alors décrites par une conductivité locale.

6
Dopage modulé d'un puits quantique: les impuretés sont dans un matériau à grand gap, les charges dans un autre à petit gap. Cette séparation des porteurs par rapport aux impuretés chargées permet d'obtenir des effets variés:
très forte mobilité High Electron Mobility Transistor Effet Hall quantifié (entier, fractionnaire) Transport cohérent, décrit par la conductance
Effet tunnel On utilise la probabilité non nulle pour un électron de traverser une barrière exemples:
semiconducteurs: GaAs / Ga1-xAlxAs / GaAs métaux: normal (Au)
magnétique (Fe) / oxyde (Al2O3) supraconducteur (Al)
Blocage de Coulomb Le confinement sur de petites dimensions entraîne une forte interaction entre porteurs
4Kà10
300KàmeV20²aF10²
nm1
nm10
0
Tk
TkCeE
hl
Ch
l
B
B
>>=
≈≈≈⇒≈≈⇒=
=
ε
εε
On peut chercher à utiliser ces effets pour réaliser, par exemple, un "transistor à un électron" ou une "mémoire à un électron". Réalisation: - îlots métalliques - effet de champ sur un puits quantique Calculateur quantique Dans une des pistes explorées, on cherche à utiliser le transfert d'un électron d'un "point quantique" à un autre en conservant la phase de sa fonction d'onde. etc…
4. Réalisation de systèmes confinés réalisation de puits quantiques en épitaxie: On empile des couches de deux ou plusieurs matériaux de propriétés électroniques différentes, avec continuité du réseau cristallin entre deux matériaux successifs (fig 6). Différents modes de croissance existent: couche par couche, en îlots, ... qu'on peut favoriser en choisissant les conditions de croissance. Ces modes de croissance seront décrits dans les premiers chapitres de la première partie.

7
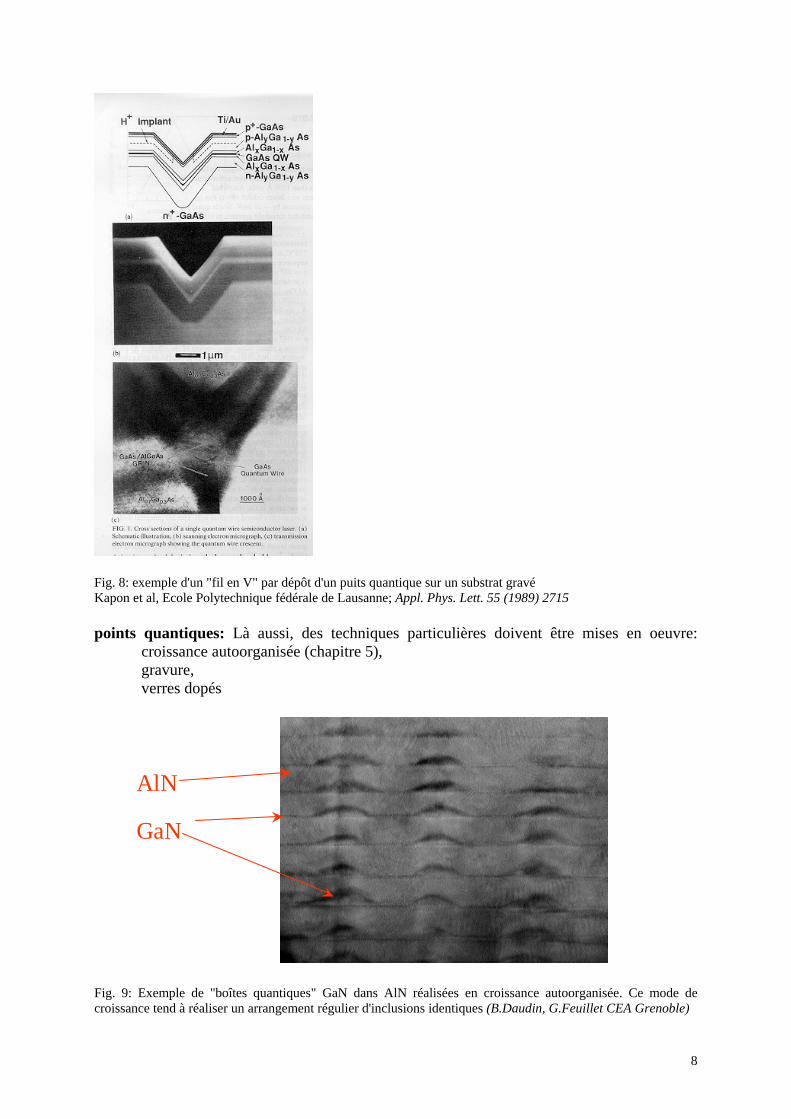
Epaisseur critique et dislocations: si les matériaux ont des paramètres de maille différents, le matériau déposé se déforme de façon élastique... tant que l'épaisseur déposée reste inférieure à une "épaisseur critique" à partir de laquelle des défauts structuraux apparaissent (chapitres 4 et 5 de la première partie). Réalisation de structures à confinement latéral: des techniques spéciales doivent être utilisées. Par exemple, le dépôt sur un substrat pré-gravé permet de réaliser un "fil quantique":
Fig. 6: image en microscopie électronique en transmission ("cross section") de puits quantiques CdTe-ZnTe. En haut: mode de microscopie conventionnelle mettant en évidence les deux matériaux; en bas: mode haute résolution mettant en évidence la continuité du réseau cristallin P.H.Jouneau et Guy Feuillet, CEA-Grenoble
Fig.7 Insertion de couches MnTe d'épaisseur croissante, dans du CdTe. Les deux matériaux ont des paramètres de maille différents. Dans les deux couches les plus fines, le matériau MnTe est déformé de façon élastique; les deux autres couches ont une épaisseur supérieure à l'épaisseur critique: des défauts compensent la différence de paramètre de maille. P.H.Jouneau et Guy Feuillet, CEA-Grenoble
8
Fig. 8: exemple d'un "fil en V" par dépôt d'un puits quantique sur un substrat gravé Kapon et al, Ecole Polytechnique fédérale de Lausanne; Appl. Phys. Lett. 55 (1989) 2715 points quantiques: Là aussi, des techniques particulières doivent être mises en oeuvre: croissance autoorganisée (chapitre 5),
gravure, verres dopés
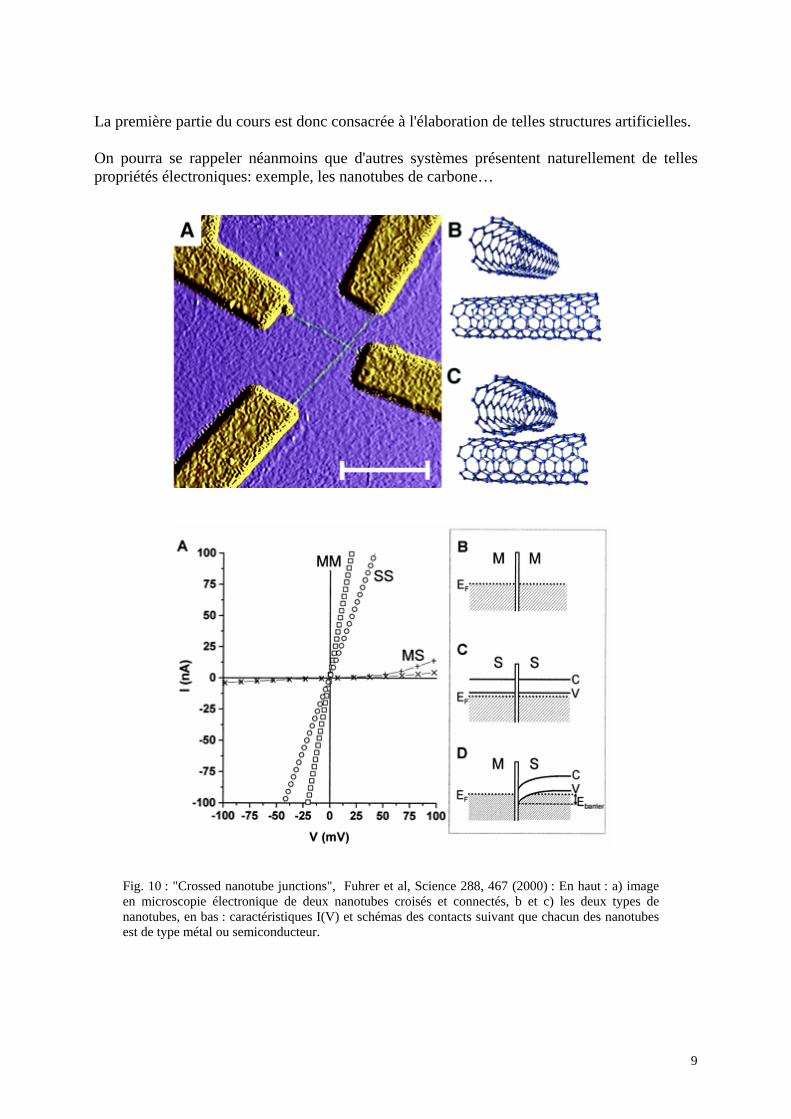
Fig. 9: Exemple de "boîtes quantiques" GaN dans AlN réalisées en croissance autoorganisée. Ce mode de croissance tend à réaliser un arrangement régulier d'inclusions identiques (B.Daudin, G.Feuillet CEA Grenoble)
AlN
GaN
9
La première partie du cours est donc consacrée à l'élaboration de telles structures artificielles. On pourra se rappeler néanmoins que d'autres systèmes présentent naturellement de telles propriétés électroniques: exemple, les nanotubes de carbone…
Fig. 10 : "Crossed nanotube junctions", Fuhrer et al, Science 288, 467 (2000) : En haut : a) image en microscopie électronique de deux nanotubes croisés et connectés, b et c) les deux types de nanotubes, en bas : caractéristiques I(V) et schémas des contacts suivant que chacun des nanotubes est de type métal ou semiconducteur.

10
Exercices Densité d'états Calculer les densités d'états 3D, 2D, 1D, 0D pour des courbes de dispersion paraboliques (données ci-dessus). Métaux, semiconducteurs On considère un échantillon de dimension L×L×L et on suppose qu'on peut le décrire par une densité d'états de type 3D. Montrer que la variation relative de l'énergie de Fermi, pour une
variation d'un électron, à température nulle, est ( )3
2
3
2)(
11LkEEdN
dEE FFDF
F
F
πρ
== .
Commenter cette valeur pour des densités d'électrons libres que vous jugerez adaptées à un métal et à un semiconducteur. On pourra prendre L = 10 nm.

11
Première partie
Elaboration
Bâti d'épitaxie par jets moléculaires, Equipe "Nanophysique et semiconducteurs" CEA-CNRS-UJF

12

13
I. Morphologie d'une surface cristalline Objectif: montrer l'influence de la structure cristalline sur la morphologie de surface. 1. Surfaces singulières
Exemples de structures cristallines: Dans ce cours, on s'appuiera le plus souvent sur deux types de structures cristallines: - la structure diamant (C, Si…) et la structure blende de zinc (ZnS, GaAs…); les liaisons sont dans les directions <111>; par définition, la direction [111] pointe de l'anion vers le cation et la direction [ ]111 du cation vers l'anion. - la structure cubique face centrée (nombreux métaux).
z=[001]
x=[100]
structure zinc-blende
structure cfc (Au…)
Zn ou Ga
S ou As
Fig.1: Mailles cubiques de la structure blende-de-zinc et de la structure cubique à faces centrées. On remarquera qu'il ne s'agit pas des mailles élémentaires: la maille cubique contiant quatre motifs élémentaires (C2, ZnS, Au). Surfaces singulières: Ce sont les surfaces d'indice faible, de haute symétrie (mais la symétrie est cependant plus basse que dans le matériau massif). Elles sont caractérisées par: - une faible densité de liaisons coupées - une anisotropie des propriétés de surface plus grande que dans le massif. On s'intéresse pour l'instant aux surfaces dites idéales, qui résultent d'une simple troncation du cristal. exemple: la surface (001) de la structure blende-de-zinc; elle peut être terminée Zn, ou S; dans ce cas, les liaisons coupées définissent la direction ]011[ . L'axe [001] est un axe d'ordre 4 dans le volume, d'ordre 2 à la surface.

14
Fig. 2: surface )001( (ici, terminée par les atomes d'anions: S dans ZnS, ou As dans GaAs) de la structure blende-de-zinc, vue suivant l'axe [001] en haut, et suivant l'axe [110] en bas. 2. Défauts sur une surface singulière De façon générale, on définit des terrasses, marches, décrochements, adatomes, adlacunes, îlots… et on aura des atomes dans la phase vapeur.
Fig.3: schéma d'une surface en cours de croissance en épitaxie par jets moléculaires (Villain et Pimpinelli) exemples de surfaces réelles observées par microscopie tunnel:
Fig. 4: surface du Si (001) Hamers et al, Ultramicroscopy 31,10 (1989):
[110]
[1-10]
[1-10]
[00-1]

15
Fig.5: surface de GaAs (001) après dépôt de 60 monocouches en épitaxie par jets moléculaires M. Sudijono et al., Surface Sci. 280, 247 (1993): 3. Surface vicinale Il s'agit d'une surface faisant un angle θ (faible) avec une surface singulière; elle est donc formée d'une succession de terrasses (plus ou moins régulières) et de marches (plus ou moins rectilignes). La structure cristalline définit la hauteur des marches élémentaires, mais on peut aussi avoir des marches empilées.
Désorientation et largeur moyenne des terrasses
l
h
La désorientation moyenne est lh
=)tan(θ
Effet de la structure cristalline: exemples des surfaces (11n) en structure blende-de-zinc (fig.6) ; elles sont obtenues à partir de la surface (001), par une rotation autour de ]011[ ; d'où
20a
h = , nlh 2tan ==θ ,
220 na
l =⇒

16
[110]
[1-10]
[1-10]
[001]
Fig.6: surface (11n) de la structure blende-de-zinc
4. Reconstruction des surfaces de semiconducteurs Les surfaces réelles de semiconducteurs sont reconstruites. Les éléments utilisés pour décrire
les reconstructions sont: - des lacunes - la formation de dimères - la relaxation (déplacement des atomes)... L'arrangement périodique de ces éléments défini une surstructure, notée ( )pn× en fonction de la nouvelle période de surface par rapport à la maille élémentaire de la surface idéale. On notera que la stoechiométrie de surface n'est en général pas la même que la stoechiométrie de volume. Méthodes d'études: pas seulement la microscopie tunnel, mais aussi la diffraction X (incidence rasante sur montage classique ou synchrotron), la diffraction d'électron (LEED Low Energy Electron Diffraction, RHEED Reflection High Energy Electron Diffraction), diverses spectroscopies des états électroniques en surface…
Fig. 7: reconstruction (2x4) de la surface (001) de GaAs; (a) image de microscopie tunnel; (b) identification des motifs; (c) interprétation de la structure (d) profil V.P.LaBella et al., Phys. Rev. Lett. 83, 2989 (1999)

17
5 Surfaces des métaux Besenbacher, Rep. Prog. Phys. 59, 1737 (1996) Elles sont souvent considérées comme plus simples, et limitées à des relaxations (en particulier déplacements perpendiculaires à la surface); mais on décrit de plus en plus d'exemples de reconstructions (rangées manquantes, …). On pourra consulter la référence indiquée. 6. Equilibre ou cinétique? Comme on le verra dans la suite, deux grandes classes de mécanismes entrent en compétition, ou en collaboration, lors de la croissance d'un solide: - l'équilibre thermodynamique - la cinétique. Il est souvent difficile de savoir ce qui a défini la morphologie d'une surface en cours d'observation: - est-ce une forme d'équilibre, déterminée par l'énergie libre et le potentiel chimique? - ou résulte-t-elle du mode de croissance, avec un aspect dynamique lié à la diffusion en surface et au collage des atomes? Réponse partielle plus loin...
Exercices 1. A partir de la figure 1, retrouver la figure 2, puis les figures correspondantes pour les surfaces )100( , (111), )111( et (110). Discuter le caractère polaire (déséquilibre entre les anions et les cations) de ces surfaces. 2. Comparer la densité de liaisons coupées sur une surface idéale de GaAs, d’orientation (001), (111), (110). 3. Sur le modèle de la figure 7, identifier les éléments de la reconstruction et la nouvelle maille élémentaire en surface.

18

19
II. Surfaces modèles et modèles de croissance But: on veut interpréter des expériences effectuées sur des surfaces réelles, mais on utilise des modèles (simplistes) qui permettent d'approcher les mécanismes physiques tout en conservant une certaine simplicité aux calculs On arrive à rendre compte des observations mais les prédictions sur l'effet de paramètres extérieurs (flux incidents, température...) sont parfois hasardeuses; ceci traduit le fait qu'on introduit des paramètres dont la signification physique (au niveau microscopique) n'est pas toujours évidente.
1. Le modèle SOS (Solid on Solid)
a. Le modèle La surface cristalline la plus simple est le voisinage de la surface (001) d'un réseau cubique simple: chaque atome est repéré par trois entiers (i,j,h) qui définissent sa position
,,, ahzajyaix === On prend une énergie de liaison El avec chacun des premiers voisins. Cela signifie que l'on gagne l'énergie El si l'on crée une liaison entre deux atomes, en supprimant deux liaisons pendantes. L'énergie de cohésion du solide est 3 El (il y a six liaisons par atome mais chaque liaison est commune à deux atomes).
Fig.1: Surface vicinale dans le modèle SOS (Hudson)
On a alors pour l'énergie des liaisons dans les différentes configurations: atome de la vapeur 0 adatome El adatome de bord de marche (ledge) 2 El atome en décrochement (kink) 3 El atome inclus en bord de marche 4 El atome inclus dans la surface 5 El atome du cristal 6 El L'énergie de formation d'un défaut se calcule sous la forme de différences entre ces énergies. Par exemple: - en présence de marches, on gagne une énergie 2 El en incorporant un adatome à un décrochement de bord de marche;

20
- sur une surface plane, faire sortir un atome pour le mettre en position d'adatome et laisser une adlacune coupe 4 liaisons et coûte 4 El; - la désorption d'un adatome (passage adatome / vapeur) demande beaucoup moins d'énergie que l'évaporation (passage d'un atome incorporé en général sur un décrochement de bord de marche, à la vapeur); on considère donc que ce passage se fait en deux étapes: du kink vers l'adatome, puis de l'adatome vers la vapeur (cf paragraphe 3.c ci dessous). - on peut aussi calculer le coût de formation d'une marche (attention : on forme deux marches à la fois…).
b. Surface d'équilibre à basse température Considérons une surface singulière. A température nulle, on n'aura pas de défauts. A basse température, on peut supposer que la surface est presque plane
10, ±= hh ji , la déviation étant due à des adatomes ou des adlacunes; la création d'une paire adatome/adlacune à partir de la surface parfaite coûtant une énergie 4 El, la probabilité p de rencontrer un adatome (une adlacune) en un site donné sera de l'ordre de )/4exp( TkE Bl− . Créer un défaut plus étendu demande une énergie plus grande et ceux-ci peuvent être négligés. La rugosité d'une telle surface se décrit par la fonction de corrélation
[ ]²)()()( rzRrzRG rrrr−+= ,
qui vaut dans ce cas ²4 ap≈ . Une telle surface, dont la fonction de corrélation reste ainsi finie à grande distance, est dite lisse. On verra plus loin (2a) un exemple de surface rugueuse.
c. Equations d'évolution Ajoutons un (faible) flux d'atomes F (nombre d'atomes par s par m²). On aura alors plus d'adatomes que d'adlacunes, et on néglige celles-ci. En pratique ce modèle est très utilisé pour effectuer des calculs numériques (Monte-Carlo) dont les résultats seront confrontés aux données expérimentales, même si celles-ci sont obtenues sur des surfaces réelles plus complexes. Dans un tel calcul, à partir d'une configuration donnée (distribution initiale d'adatomes sur la surface), on calculera l'évolution en écrivant que pendant un intervalle de temps tδ , il peut se produire: - l'arrivée d'un adatome (probabilité tFapF δ2= ), - la désorption d'un adatome (probabilité desp par adatome), - le saut d'un adatome de son site à un site voisin (probabilité sautp par adatome), - et l'incorporation, rappelée ici par le terme pincorp. A partir de ces mécanismes, en considérant un ensemble de telles configurations, on peut écrire une équation d'évolution de la probabilité jin , d'avoir un adatome sur le site ),( ji :
[ ])()()()()()()()(
)()()(
,1,,1,,,1,,1
,,,
tntntntntntntntnp
ptnpptnttn
jijijijijijijijisaut
incorpjidesFjiji
−+−+−+−+
−−+=+
−+−+
δ
le problème difficile étant de tenir compte de l'incorporation, problème qu'on abordera plus loin. Cette équation peut se résoudre soit par un calcul numérique, soit par une "approximation continue" (voir paragraphe 2b dans ce chapitre).

21
2. Modèle continu
a. Surface rugueuse à haute température Dans cette approche, la surface se décrit par la donnée de la fonction ),( yxz ; dans le paragraphe précédent, où la surface est presque plane, on aurait 0),( zyxz = avec une probabilité ),(1 yxn− , et azyxz += 0),( avec une probabilité ),( yxn , où a est la hauteur de marche (on peut ajouter un terme équivalent azyxz −= 0),( pour les adlacunes). Mais à haute température les fluctuations sont grandes et la surface s'écarte beaucoup de la surface plane. L'hypothèse essentielle est alors que la fonction ),( yxz est continue.
On peut écrire l'aire de l'élément de surface correspondant à un élément d'intégration dydx :
dydxyz
xzdS
22
1 ⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎠⎞
⎜⎝⎛∂∂
+=
Si l'on suppose en outre que l'énergie de la surface est donnée par une énergie par unité de surface σ uniforme et isotrope (ce serait le cas d'un liquide, et on parlerait alors de tension superficielle), alors
dydxyz
xzL
dydxyz
xz
dydxyz
xzdSE
LL∫∫
∫∫
∫∫∫∫
× ⎥⎥⎦
⎤
⎢⎢⎣
⎡⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎠⎞
⎜⎝⎛∂∂
+=
⎥⎥⎦
⎤
⎢⎢⎣
⎡⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎠⎞
⎜⎝⎛∂∂
+≈
⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎠⎞
⎜⎝⎛∂∂
+==
22
22
22
21²
21
211
1
σσ
σ
σσ
C'est l'énergie d'une surface particulière, dont la morphologie est décrite par une fonction z(x,y) donnée. On va chercher la valeur moyenne de E pour une distribution thermique de telles surfaces. Pour cela, on va d'abord chercher les modes propres. Ils s'obtiennent par une transformation de Fourier:
⎟⎟⎠
⎞⎜⎜⎝
⎛=⎟⎟
⎠
⎞⎜⎜⎝
⎛=
yx
rqq
qy
x rr , et ).exp(),( rqizyxzq
qrr
rr∑= , d'où ).exp(),( rqizqi
xyxz
qqx
rrr
r∑=∂
∂ ; l'énergie
pour une configuration qzr , définie par la donnée de tous les qzr , est alors
∑+=q
qzqLcteEr
r ²²2²σ .
Cette formule décrit encore l'énergie d'une surface particulière, dont la morphologie est décrite maintenant par la donnée des zq. La probabilité d'une configuration qzr est le produit
dS
dx
dxxz
∂∂
z(x,y)

22
des probabilités pour chaque mode qzr . En appliquant à ces modes propres indépendants la statistique de Boltzmann, on calcule la valeur moyenne sur toutes les configurations possibles:
²²1)/exp()/exp(²²
qTk
LdzTkEdzTkEzz B
qBqqBqqq σ=−−= ∫∫ rrrrrr
(c'est le théorème d'équipartition) que l'on peut utiliser pour calculer enfin la fonction de
corrélation [ ] [ ]∫ −+=−+= ²)()(1²)()()( 22 rzRrzrd
LrzRrzRG rrrrrrr
(on prend deux
moyennes, une sur une surface donnée par l'intégrale double sur r, et l'autre sur la distribution de surfaces):
[ ] ).exp(1).exp()()( rqiRqizrzRrzq q
rrrrrrrr r −=−+ ∑
et, puisque ( ) ',23 .'exp qqLrqqird rr
rrr δ=−∫
[ ] [ ]
( )∫ ∫
∑
∑
−⎟⎠⎞
⎜⎝⎛≈
−=
−−= −
aB
qB
Rqiq
Rqiq
qqRqdqdL
LTk
qRq
LTk
eezRG
/1
0
2
0
2
..
²coscos1
2²2
²).cos(1
²2
11²)(
π
σθθ
π
σr
rr
r
rrr
rr
r
L'intégrale ( )[ ] θθπ
dqR∫ −2
0
coscos1 se trouve dans les tables, se calcule numériquement (figure
ci-contre) ou s’évalue avec un peu de réflexion : elle est de l'ordre de (qR)² si qR est petit (il suffit de faire un développement limité du cosinus: cf la droite sur la figure) et constante si qR est grand (le cosinus se moyenne). Il reste donc
∫ −=≈a
R
aRq
qdq/1
/1
)Ln()Ln(²
d'où )Ln()( RTk
RG B
σ≈
r.
La fonction de corrélation diverge à longue distance: la surface est dite rugueuse. On a donc: - à basse température, une surface lisse, qu'on décrira dans le modèle "TLK", "terrace-ledge-kink", c'est-à-dire des terrasses séparées par des marches comportant des décrochements (1er paragraphe). - à haute température, une surface rugueuse, qu'on décrira par une fonction ),( yxz continue. Entre les deux se produit la "transition rugueuse".
b. Modèle intermédiaire: équations d'évolution des adatomes sur une terrasse On considère la densité d'adatomes ²/),( , anyx ji=ρ pour jayiax == , . Un modèle continu décrivant l'évolution de ρ est obtenu comme la limite du modèle précédent lorsque 0→tδ et
0→a .
0.1 1 100.01
0.1
1
10
Ln (i
ntég
rale
)
Ln (qR)

23
ttatnttn jiji
∂∂
→−+ ρ
δδ²
)()( ,, ,
[ ] ⎥⎦
⎤⎢⎣
⎡∂∂
+∂∂
→−+−+−+− −+−+ ²²
²²²
²1
,1,,1,,,1,,1 yxannnnnnnn
a jijijijijijijijiρρ ,
d'où, en posant
Fta
pF =δ²
, τρ
δ=
tanp jides
², ,
tp
aD ss δ
²= ,
l'équation d'évolution de );,( tyxρ :
incs dt
dDFt ⎥⎦
⎤⎢⎣⎡+∆+−=
∂∂ ρρ
τρρ .
On a donc une description continue. On peut aussi définir le courant: nombre de particules franchissant une ligne par unité de longueur et de temps. Pendant le temps tδ et à travers une longueur a, il passe
taj δ particules qui correspondent aux sauts ( )iis nnp −−1 . D'où
( ) ( )( )x
paxaxpataj ss ∂∂
−=−−=ρρρδ 32 soit
xDj s ∂
∂−=
ρ .
3. Exemples d'applications du modèle SOS
a. Choix de paramètres réalistes Ces modèles sont utilisés couramment et confrontés à des mesures expérimentales sur des surfaces réelles, telles que les surfaces de semiconducteurs, bien que celles-ci soient bien plus complexes. On aura couramment recours à des paramètres tels que: - pour le coefficient de diffusion, ( )TkmEEaD BlDs /)(exp² +−=ν où:
-11312 s10102 −≈≈ hTk Bν est une fréquence de vibration caractéristique
DE représente la barrière à franchir pour qu'un adatome isolé saute d'un site au voisin, de l'ordre de 1 eV
lE est l'énergie de liaison avec les voisins (fraction de l'eV) m est le nombre de voisins (m=0 pour un adatome isolé, et on reprend les idées du modèle SOS décrit plus haut)
- pour la désorption )/exp(0 TkE Bdesττ = conduisant à une probabilité de désorber qui peut être négligeable (c'est le cas de l'épitaxie de GaAs), ou presque comparable à la probabilité de diffuser (c'est le cas de l'épitaxie de CdTe). On voit que le terme de diffusion, tel qu'il est écrit ci-dessus, s'applique non seulement à la diffusion d'un adatome (m=0), mais décrit aussi l'incorporation. Celle-ci se fait par incorporation en bord de marche, ou par nucléation sur les terrasses (deux adatomes se rencontrent et forment une paire, qui évolue en triplet, etc). Plus un agrégat est gros, et plus il est stable: on définira alors une taille critique de nucléation, qui est la taille à partir de laquelle un agrégat est pratiquement stable.
b. Résultats La figure 2 donne les résultats d'un calcul simple, où le minimum de paramètres ont été introduits. On peut modifier ce genre de calcul pour tenir compte de structures plus complexes: ainsi la dissymétrie de la surface (001) du GaAs crée une différence entre les marches parallèles aux

24
directions <110> (figure 3) et une anisotropie de la diffusion, deux effets qui peuvent être introduits "de force" dans les paramètres du modèle (figure 4).
4. Modèle BCF C'est LE modèle classique de la croissance cristalline (Burton, Cabrera, Frank). On applique ici le calcul à une surface avec des marches dans une seule direction, et pas d'autres défauts (Villain et Pimpinelli).
Fig. 2: Calcul Monte Carlo: dépôt d'une fraction de monocouche à 500K (a), 600K (b), 680K (c), pour des paramètres adapté à GaAs Clarke et Vvedenskii, J. Appl. Phys. 63, 2272 (1988) A basse température, on observe la nucléation sur les terrasses; à haute température, les atomes diffusent jusqu'aux marches et s'y incorporent.
Fig. 3 :Schéma des bords de marches à la surface (001) de GaAs Shitara et al., Phys. Rev. B 46, 6825 (1992)
Fig. 4: Effet de l'anisotropie de l'incorporation aux bords de marche dans GaAs; Shitara et al., Phys. Rev. B 46, 6825 (1992). Comparer aux figures 4 et 5 du chapitre 1.

25
L'équation d'évolution continue donnée plus haut, réduite à une dimension x, s'écrit:
incs dt
dx
DFt ⎥⎦
⎤⎢⎣⎡+
∂∂
+−=∂∂ ρρ
τρρ
²² ;
On considère une terrasse de largeur l , et on place l'origine des x au milieu de cette terrasse. On suppose que toute l'incorporation se fait en bord de marches ("croissance par avancée de marches"), ce qu'on décrit: - en annulant le terme d'incorporation sur la terrasse (pas de nucléation sur les terrasses) - en imposant au voisinage d'une marche 0)2/()2/( ρρρ ==− ll , la densité d'équilibre avec la marche. Ce terme reste un peu mystérieux, on y reviendra plus loin. On peut dire néanmoins que )/exp(0 TkE Bak−−≈ρ , où akE − est l'énergie nécessaire pour faire passer un atome d'une position de décrochement en bord de marche (kink) à la position d'adatome. - en écrivant que la vitesse de croissance du cristal, l// aVdtdz = , où la vitesse de la marche dans la direction x est la somme des courants de part et d'autre:
xDxj
jjaV
s ∂∂
−=
⎥⎦⎤
⎢⎣⎡ +−−−=
ρ)(
)02
()02
(² ll
On détermine donc la densité sur la terrasse, en régime permanent, comme solution de
0²
²=
∂∂
+−=x
DFdtd
sρ
τρρ , vérifiant les conditions aux limites en 2/l± , ce qui conduit à
)()2/cosh(
)cosh()( 0 τρκκτρ FxFx −+=l
,
où on a posé τκ sD1² = ; d'où l'on déduit le courant:
)sinh()2/cosh(
)( 0 xF
Dxj s κκ
τρκ
l
−−=
et la vitesse de la marche entre deux terrasses de largeurs l et 'l [ ])2/tanh()2/tanh()(² 0
−+ +−= ll κκρτκ FDaV s .
0 2 4
V
κl-0.4 -0.2 0.0 0.2 0.4
F=0
κl=1
κl=10
xl
ρ0
Fτ
Fig. 5: Densité d'adatomes (à gauche) et vitesse d'avancée des marches (à droite)
On en déduit la vitesse de croissance du cristal (avancée de la surface dans la direction z):

26
⎟⎠⎞
⎜⎝⎛⎟
⎠
⎞⎜⎝
⎛ −==2
tanh203 l
ll
κκτ
ρFaVa
dtdz .
Commentaires: - si τsD/l est petit: tous les atomes arrivant sur la terrasse s'incorporent à une marche voisine. Alors 0ρρ = partout et 0ρ est donc la densité d'adatome à l'équilibre solide-vapeur. On en donnera une expression au prochain chapitre. - si τsD/l est grand: seuls les atomes arrivant à une distance inférieure à la longueur de diffusion atteignent la marche, les autres "désorbent". Domaine de validité: le modèle BCF tel qu'il est décrit ici est applicable en l'absence de nucléation sur les terrasses; on montre qu'une telle nucléation se produit si ( ) 6/1)/ FDs>>l .
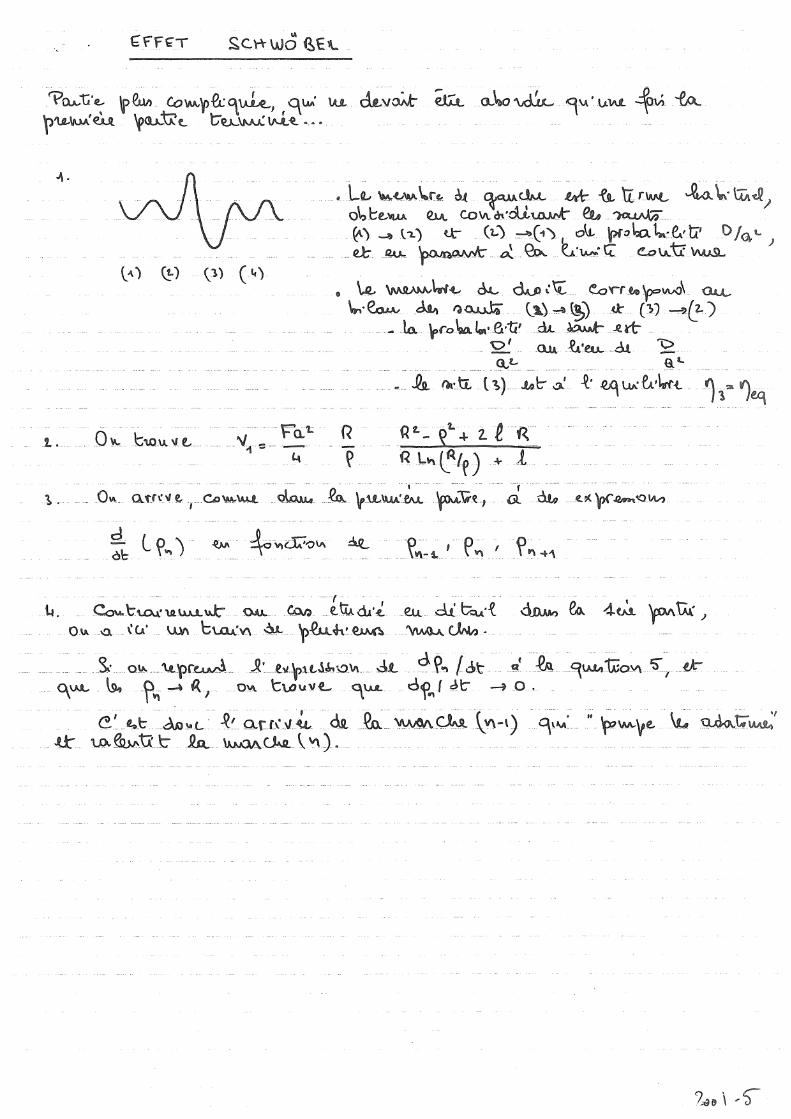
Exercices 1. Modèle SOS On considère un cube de côté L = na (n entier) d'un solide SOS de longueur de liaison a. Dénombrer le nombre total de liaisons formées, et en déduire l'énergie de cohésion par atome, et l'énergie de la surface (001) (par liaison pendante, puis par unité d'aire). 2. Effet Schwöbel: Dans le modèle BCF ci-dessus, on a supposé que tout adatome arrivant sur une marche (que ce soit par la terrasse du dessus ou par celle du dessous) est susceptible de s'incorporer à la marche et donc de la faire avancer. A la surface de nombreux matériaux, le franchissement d'une marche est difficile et disymétrique ("effet Schwöbel"). Que devient le modèle BCF en cas d'effet Schwöbel extrême (tel que l'adatome arrivant à une marche par la terrasse du dessus est réfléchi)? 3. Montrer de façon qualitative que l'effet Schwöbel tend à régulariser la largeur des terrasses pendant la croissance (on évaluera la vitesse d'avancée d'une marche en fonction de la taille de la terrasse adjacente). 4. Allez voir des simulations de croissance (couche par couche, par déplacement de marches, en « ALE ») à l’adresse http://theorie.physik.uni-wuerzburg.de/~ahr/AB/
Fig. 6: potentiel vu par les adatomes au voisinage d'une marche monoatomique schématisée en bas. En haut (ligne continue), on tient compte de l'effet Schwöbel; au milieu (ligne pointillée), on l'ignore.

27
III. Formes d'équilibre L'équilibre entre un solide et la vapeur est décrit, en première approximation, en ignorant que l'interface entre les deux phases (la surface du solide) est un objet qui peut être complexe: une surface donnée a priori n'est pas nécessairement en équilibre avec le solide ou la vapeur, ni même en équilibre interne. C'est la condition de cet équilibre qu'on veut établir.
1. Equilibre solide-vapeur Point de départ: à l'équilibre, on a l'égalité des potentiels chimiques de la vapeur et du solide
sv µµ = La vapeur est décrite comme un gaz parfait:
⎟⎟⎠
⎞⎜⎜⎝
⎛−= 3Ln
λµ
pTk
Tk BBv
où ( ) 2/12 Tmkh Bπλ = est la longueur d'onde de de Broglie. Le solide est décrit, par exemple, dans le modèle d'Einstein: Pour chacun des N atomes du solide, on a une énergie U0 et un oscillateur harmonique à trois degrés de liberté. Pour les oscillateurs, l'énergie libre )Ln(ZTkF B−= se calcule par
⎥⎦
⎤⎢⎣
⎡−−
⎥⎦
⎤⎢⎣
⎡−
=⎥⎦
⎤⎢⎣
⎡+−= ∑∞
=
Tkh
Tkh
TkhnZ
B
Bn
B ν
νν
exp1
21exp
)21(exp
0
d'où, pour le solide ( ) >−−<+><+== )/exp(1Ln33/ 2
10 TkhTkhUNF BBs ννµ
et si on prend tous les oscillateurs identiques (modèle d'Einstein), dans la limite haute température ( νhTk B >> ):
⎟⎟⎠
⎞⎜⎜⎝
⎛++=
TkhTkhU
BBs
ννµ Ln323
0
On définit l'énergie de sublimation à T=0 ( 0LTk B << ), νhUL 23
00 −−= . La condition
sv µµ = permet de calculer la pression de vapeur saturante
( )( ) ⎟⎟
⎠
⎞⎜⎜⎝
⎛−=
TkL
Tkmp
BBs
02/1
2/3
exp²2 νπ .
C'est une propriété du matériau, indépendamment de la surface considérée! Cela signifie que dans cette description, la surface (ses terrasses, marches, adatomes...) n'est pas arbitraire: elle est en équilibre avec le solide et la vapeur. Valeurs typiques:
Elément L0 (eV) ν (THz) hν (meV) Ag (fcc) 2.95 eV 4 16 Fe (bcc) 4.28 eV 11 43
Si 4.63 eV 15 59 Ge 3.83 6 24 Ar 0.085 1 4

28
Le modèle de gaz parfait permet de calculer le flux de particules passant à travers un élément de surface Sδ : la densité d'atomes dans la vapeur est Tkp B/ , et on doit prendre celles qui se trouvent dans un volume tS δδ
rr.v . En coordonnées cylindriques, on a
( ) 2/12/
0 0
22
0
2/
0 0
32
0
2)2/²exp()sin(
)2/²exp()sin()cos(
Tmkp
Tkp
dTkmdd
dTkmddF
BBB
B
πθθϕ
θθθϕ
ππ
ππ
=−
−=
∫ ∫∫
∫ ∫∫∞
∞
vvv
vvv
On peut utiliser cette expression pour calculer l'évaporation à partir d'une face d'un cristal. L'évaporation se fait par désorption d'adatomes: soit 0ρ la densité d'adatomes à l'équilibre et τ/1 la probabilité de désorption (par unité de temps) d'un adatome; on suppose que solide, marches, terrasses et vapeur sont en équilibre. L'équilibre entre les adatomes désorbés et les atomes arrivant (on suppose que le coefficient de collage est 1, c'est-à-dire que chaque atome incident est incorporé) s'écrit:
( )( ) ⎟⎟
⎠
⎞⎜⎜⎝
⎛−==
TkLmTk
Tmkp
BB
B
s 032
22/1
0 exp42 hππτ
ρ
Si la densité d'adatomes est en équilibre avec des marches, )/exp(0 TkE Bak −−≈ρ , où akE − est l'énergie nécessaire pour faire passer un atome d'une position de décrochement en bord de marche (kink) à la position d'adatome. La probabilité de désorption τ/1 a alors pour énergie d'activation akEL −−0 , qui correspond bien à la désorption des adatomes. Dans le modèle SOS, lEL 30 = (El énergie par liaison, il y a six liaisons par paire d'atome),
lak EE 2=− et lak EEL =− −0 . On peut revenir au modèle Burton-Cabrera-Frank; celui-ci montre que: - si la distance entre marches est faible devant la longueur de diffusion, 0ρρ = sur toute la terrasse et l'évaporation se fait avec le flux calculé par la pression de vapeur saturante; - dans le cas contraire la densité entre deux marches est plus faible que 0ρ et il faut prendre en compte les échanges entre les différentes parties de la surface. La vitesse de croissance ou de sublimation d'une surface donnée a priori dépend donc de la densité de marches. On peut aussi appliquer ce calcul au "flux moléculaire" issu d'une cellule dans un bâti d'épitaxie par jets moléculaires. Pour avoir le flux incident sur le substrat placé à une distance SL δ>> de l'embouchure de la cellule, on doit tenir compte de l'angle solide sous lequel la cellule est vue. Des calculs numériques sont nécessaires pour tenir compte de la géométrie réelle.
2. Rôle de la surface: cas d'un liquide On va mettre en évidence deux paramètres importants: l'énergie de la surface et sa courbure.
a. Goutte isolée (Gibbs-Thomson) On suppose que la surface est caractérisée par une énergie proportionnelle à son aire, isotrope:
SE σ= . Ceci définit l'énergie de surface (appelée tension superficielle pour un liquide), σ.

29
La formation d'une goutte de rayon R contenant N atomes avec une densité n entraîne une variation du potentiel thermodynamique
33
4)(²4)( RnRNSG vsvs πµµπσµµσ −+=−+=∆ La dérivée par rapport à R s'annule pour
( )svnR
µµσ−
=2 .
- sur une surface plane à l'équilibre, vs µµ = . Hors équilibre, on définit la sursaturation ( )sBsv ppTk /Ln)( =−=∆ µµµ où p est la pression dans la vapeur et ps la pression de
vapeur saturante (qui donnerait l'équilibre entre la surface plane et la vapeur); - une goutte de rayon donné est en équilibre avec la vapeur à une valeur de sursaturation donnée par la relation de Gibbs-Thomson: - dans une assemblée de gouttes inhomogènes en taille, il y aura transport dans la vapeur, du voisinage des petites gouttes vers les grosses ("mûrissement d'Ostwald").
b. Goutte sur un substrat plan Un calcul direct donne: - l'aire de l'interface )²sin( απ RAi = - l'aire de la surface extérieure )cos1²(2 απ −= RAd
- le volume 3
coscos32 33 ααπ +−
= RV
On a, en notant sdi σσσ ,, les énergies superficielles de l'interface, du dépôt et du substrat, et n la densité volumique:
)()( svisidd nVAAG µµσσσ −−−+=∆ . On doit annuler les dérivées partielles RG ∂∆∂ / et α∂∆∂ /G . Tout calcul fait, on en déduit R et la relation de Young: ασσσ cosdis =− qu'on peut obtenir directement: une force s'exerçant sur la ligne de raccordement est obtenue en dérivant chacune des énergies de surface ou d'interface par rapport à la position de la ligne de raccordement, la résultante de ces trois forces suivant le plan du substrat est nulle à l'équilibre.
3. Solide: rôle de l'anisotropie Deux modifications par rapport au liquide: - si on applique une contrainte, on modifie l'énergie par unité de surface. Cet effet, qui n'existe pas pour les liquides (leur surface s'adapte à toute déformation), conduit à définir des "efforts de surface"; on n'en parlera pas ici mais il peuvent jouer un rôle significatif (voir Villain et Pimpinelli) - l'énergie de surface est en général anisotrope
α
α
dépôt
substrat
30
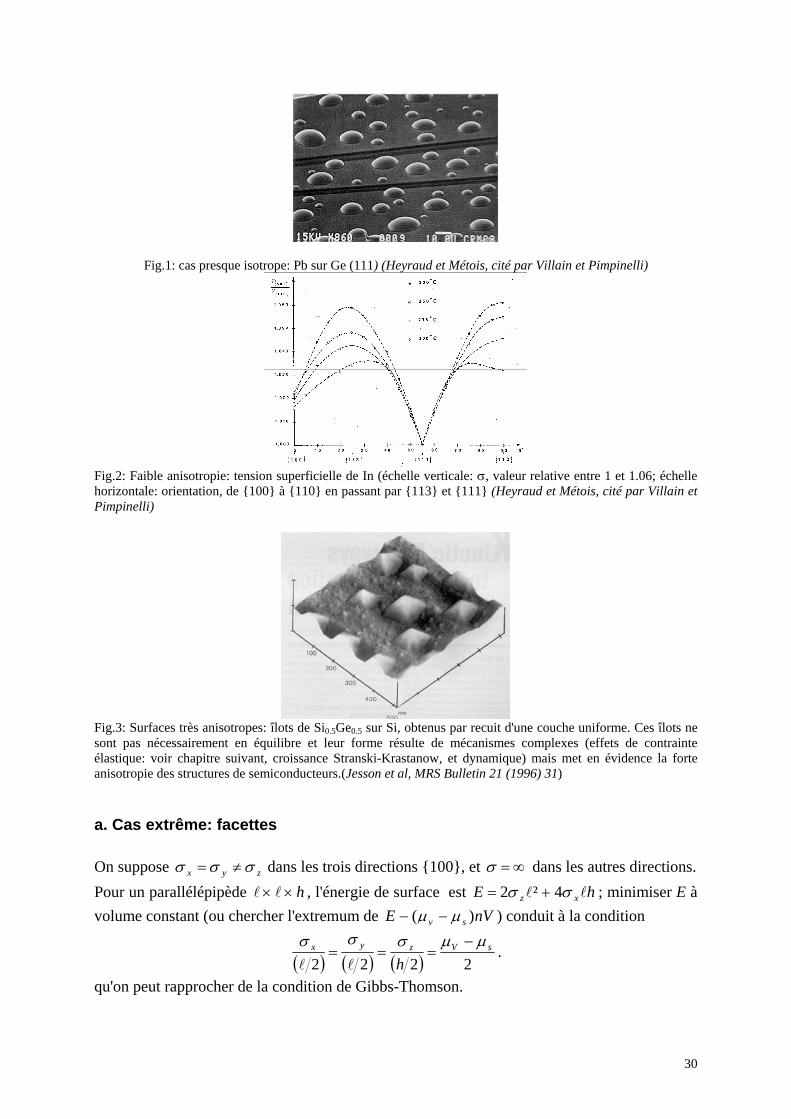
Fig.1: cas presque isotrope: Pb sur Ge (111) (Heyraud et Métois, cité par Villain et Pimpinelli)
Fig.2: Faible anisotropie: tension superficielle de In (échelle verticale: σ, valeur relative entre 1 et 1.06; échelle horizontale: orientation, de 100 à 110 en passant par 113 et 111 (Heyraud et Métois, cité par Villain et Pimpinelli)
Fig.3: Surfaces très anisotropes: îlots de Si0.5Ge0.5 sur Si, obtenus par recuit d'une couche uniforme. Ces îlots ne sont pas nécessairement en équilibre et leur forme résulte de mécanismes complexes (effets de contrainte élastique: voir chapitre suivant, croissance Stranski-Krastanow, et dynamique) mais met en évidence la forte anisotropie des structures de semiconducteurs.(Jesson et al, MRS Bulletin 21 (1996) 31)
a. Cas extrême: facettes On suppose zyx σσσ ≠= dans les trois directions 100, et ∞=σ dans les autres directions. Pour un parallélépipède h×× ll , l'énergie de surface est hE xz ll σσ 4²2 += ; minimiser E à volume constant (ou chercher l'extremum de nVE sv )( µµ −− ) conduit à la condition
( ) ( ) ( ) 2222sVzyx
hµµσσσ −
===ll
.
qu'on peut rapprocher de la condition de Gibbs-Thomson.

31
De façon plus générale, lorsque les surfaces de faible énergie superficielle ont des directions variées, on arrive à la condition
)(2
...2
2
1
1sv
nhhh
µµσσσ
α
α −===
où α désigne la direction et hα est la distance au centre. On utilise alors la construction de Wulff, qui consiste à tracer dans chaque direction, à partir d'un point dit "point de Wulff", un segment proportionnel à σ, et le plan perpendiculaire à l'extrémité de ce segment.
La forme obtenue est une forme d'équilibre. La sursaturation de la vapeur précise la taille.
b. Modèle continu: potentiel chimique local Dans le cas d'anisotropie faible, à haute température (au dessus de la transition rugueuse) on décrit la surface par une fonction continue dérivable ),( yxz . On a alors: - le volume ∫∫
×
=LL
dxdyyxzV ),(
- l'énergie de la surface dxdyzzdSE yLL
xLL
)','(∫∫∫∫××
== ϕσ où l'on a posé
)²'()²'(1)','()','(
/'
yxyxyx
x
zzzzzz
xzz
++=
∂∂=
σϕ
où on note la dépendance de l'énergie superficielle σ par rapport à l'orientation de la surface, définie par les dérivées. On cherche un extremum de nVEG sv )( µµ −−=∆ , en supposant que la surface
),( yxz varie d'un écart ),( yxzδ , par hypothèse nul au voisinage des extrémités. Alors
dxdyzz
zz
dxdyznG yy
xx
∫∫ ∫∫⎥⎥⎦
⎤
⎢⎢⎣
⎡
∂∂
+∂∂
+∆−=∆ ''
''
)( δϕδϕδµδ
( sv µµµ −=∆ et n=densité d'atomes) La deuxième intégrale s'intègre par parties
∫∫∫ ⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
∂∂
−⎥⎦
⎤⎢⎣
⎡∂∂
=∂∂
∂∂
=∂∂ L
x
L
x
L
x
L
xx
dxzzx
zz
dxzxz
dxzz 0000 '''
''
δϕδϕδϕδϕ
d'où
dxdyzyzx
nzGyx
∫∫⎥⎥⎦
⎤
⎢⎢⎣
⎡
∂∂
∂∂
+∂∂
∂∂
+∆−=∆''
)( ϕϕµδδ .
z(x,y)
z+δz

32
Cette quantité s'annule pour toute variation ),( yxzδ si:
⎥⎥⎦
⎤
⎢⎢⎣
⎡
∂∂
∂∂
+∂∂
∂∂
−=yx
sv zyzxn ''1 ϕϕµµ
C'est la formule de Herring-Mullins, où le membre de droite définit un potentiel chimique local. Que signifie cette formule? Dans le cas d'une surface relativement régulière, ces quantité peuvent se calculer par un développement limité. On va le faire au voisinage d'une surface de haute symétrie pour simplifier les calculs. En posant yx zz '/'/' ∂∂=∂∂= σσσ ²'/²²'/²'' yx zz ∂∂=∂∂= σσσ , et 0''/² =∂∂∂ yx zzσ , on a:
[ ] [ ] )²'()²'(1)²'()²'('')''(' 21
21
yxyxyx zzzzzz ++++++≈ σσσϕ
d'où xx
zz
')''(''
σσσϕ++=
∂∂ et ( ) xx
x
zzx
'''''
σσϕ+=⎟⎟
⎠
⎞⎜⎜⎝
⎛∂∂
∂∂ .
On définit la rigidité de surface ''~ σσσ += . Attention, la dérivée est par rapport à xz ∂∂ / , c'est-à-dire en fait par rapport à l'angle θ définissant l'inclinaison de la face. La formule de Herring-Mullins devient donc
Rnz
n sxxsσµσµµ~2''~2
±=−= , avec 2
2~
θσσσ
dd
+= .
Dans le dernier terme, le signe + correspond à une surface convexe et le signe – à une surface concave.
La condition d'équilibre vµµ = devient nRsvσµµµ~2
±=−=∆ , c'est-à-dire que la rigidité de
surface a remplacé l'énergie de surface dans la formule de Gibbs-Thomson. De plus, on a l’expression pour une courbure quelconque (convexe ou concave suivant le signe).
c. Construction de Wulff: modèle continu Pour simplifier, on fait le raisonnement en coordonnées polaires à deux dimensions (c'est-à-dire que le calcul s'applique en fait à un îlot délimité par une marche fermée). On traduit simplement la construction de Wulff discrète en une version continue en θ, qu'il suffit d'expliciter en coordonnées polaires: On trace, à partir du point de Wulff O et dans la direction d'angle polaire θ, un segment OH proportionnel à σ
OH=σ ur où ur est le vecteur unité d'angle θ (de composantes θθ sin,cos ).
Puis on trace la perpendiculaire en H un point M sur cette perpendiculaire est défini par HM=α uθ, où uθ est le vecteur de composantes
θθ cos,sin− et α est arbitraire. "Couper ce qui est à l'extérieur" revient alors à prendre la courbe enveloppe de ces perpendiculaires
On détermine α(θ) pour que la droite HM soit tangente à la courbe: il faut que dM/dθ soit suivant uθ.
Il reste à expliciter cette dernière condition en utilisant les relations classiques sur les courbes en coordonnées polaires, les abscisses curvilignes et la courbure (colonne de gauche) appliquées à la construction de Wulff (colonne de droite):

33
θθu
u=
dd r , rd
du
u−=
θθ ( ) ( ) ( ) θθ ασασασ
θθuuuuOM '' ++−=+= rrd
dd
d
d'où 'σα = et OM=σ ur+σ' uθ. Pour une courbe définie par une expression paramétrique ( )θOM l'abscisse curviligne ( )θs est définie par
TOMθθ d
dsd
d= où T est le vecteur tangent
Ici, θuT = et
σσσθ
~"=+=dds
La courbure est calculée par
Rdsd 1.2
2
−=NOM où N est le vecteur
normal
Ici, ruN = et ( ) θσσθ
uOM "+=d
d ,
d'où R=σ~
La construction de Wulff conduit donc à une courbe qui reproduit bien la condition de Gibbs-Thomson, et dont l'équation s'écrit OM=σ ur+σ' uθ. Commentaires: - La courbe obtenue est arrondie mais favorise les directions de faible énergie de surface. - En cas de forte anisotropie, on voit apparaître des facettes.
d. Différents types de croissance épitaxiale La relation de Young, ασσσ cosdis =− définit les conditions de mouillage d'un dépôt sur un substrat, et la morphologie du dépôt. Croissance de type Frank-van-der-Merwe: Si ids σσσ +> , on ne peut pas trouver d'angle vérifiant la condition de Young; en fait, l'inégalité exprime que le dépôt minimise son énergie s'il couvre au maximum le substrat (mouillage parfait). Dans ce cas, le dépôt a tendance à se faire couche par couche (on finit la couche n avant de commencer la couche n+1). Evidemment, il y a aussi un aspect cinétique: ceci est vrai seulement si les adatomes diffusent assez vite à la surface pour s'incorporer aux bords d'îlots (voir ci-dessous). Croissance de type Volmer-Weber: Si ids σσσ +> , le dépôt forme des îlots. Si l'énergie de surface est isotrope, la forme des îlots est caractériséepar l'angle α de la relation d'Young. Si l'énergie de surface est anisotrope, il faut bien sûr réexaminer la relation d'Young. Dans le cas de facettes, il suffit de modifier le calcul du paragraphe 3a. Par exemple, dans le cas d'un parallélépipède, l'énergie devient
hE xsid lll σσσσ 4²)(² +−+=
De façon générale, la forme d'équilibre sur un substrat se déduit de la forme d'équilibre dans le vide par une troncation dans la direction i de l'interface (Kaishew 1952; voir Müller et Kern, J. Crystal Growth 193 (1998) 257):
)(2
...2
2
1
1sv
i
si nhhh
µµσσσσ
−====−
34
4. Cinétique. Les formes d'équilibre sont rarement atteintes. La forme réelle fait intervenir la dynamique de la croissance: diffusion de surface, incorporation en bord de marche, vitesse de croissance des facettes… On a vu un exemple du rôle de la nucléation sur les terrasses, puis de l'anisotropie de l'incorporation en bord de marche dans le cas de GaAs (001) au chapitre précédent. Dans le cas de croissance de type Frank- van der Merwe, on mesure en cours de croissance des oscillations de la rugosité de surface (oscillations dans l'intensité de réflexion d'un faisceau d'électron). Dans une interprétation simplifiée de ces oscillations, la réflectivité est maximum sur la surface initiale parfaitement lisse, diminue lorsque des îlots de croissance sont nucléés, est minimale lorsque la monocouche en formation a un taux de couverture 1/2, puis ré-augmente lorsqu'elle se complète. La période de ces oscillations correspond donc à la croissance d'une monocouche, et on suppose que la décroissance de l'intensité réfléchie est proportionnelle à la densité de marches à la surface. Cependant, l'intensité de ces oscillations diminue au cours du temps, ce qui montre qu'on crée des îlots de la couche n+1 avant de terminer la couche n, simplement parce que la diffusion des adatomes en surface est finie.
Fig.4: Oscillation de l'intensité réfléchie pour un faisceau d'électrons en incidence rasante ("oscillations RHEED") lors de la croissance de type Frank-van der Merwe
Cette nucléation d'îlots "bidimensionnels" (d'épaisseur 1 monocouche) n'existe pas si la densité de marche est suffisante: on n'observe alors pas d'oscillations. C'est le cas d'une surface vicinale (la croissance a lieu par avancée de marches), ou d'une surface singulière où la croissance a lieu depuis un certain temps (les oscillations se sont amorties). On observe les oscillations après un arrêt de croissance qui a "nettoyé" les terrasses. Les îlots se forment alors par interaction entre adatomes diffusant sur la terrasse. La "taille critique" est celle du plus petit agrégat stable.
Lors de la croissance d'îlots épais, on peut utiliser une "construction de Wulff dynamique": la forme des îlots est obtenue en traçant, à partir du point O, un segment proportionnel à la vitesse de croissance d'une surface de direction donnée. On suppose dans ce cas que la diffusion à la surface est grande. On voit rapidement que, pour un cristal convexe, par exemple des îlots :

35
- lors de la croissance, ce sont les facettes dont la vitesse de croissance est faible qui apparaissent; on finit par avoir une forme « self-similaire », qui se conserve pendant la croissance ; - lors de l'évaporation, ce sont celles dont la vitesse de sublimation est forte (mais dans ce cas la forme obtenue dépend bien sûr de la forme initiale!). Par contre, pour la croissance à partir d’une forme concave (trou, sillon : voir les « fils quantiques » du premier chapitre) ce sont les facettes de forte vitesse de croissance qui subsistent. Flux et gradient de potentiel chimique A l'équilibre, le potentiel chimique est uniforme. Si le potentiel chimique varie, le gradient de potentiel gouverne le mouvement des particules (par exemple, adatomes): dans le cas d'un mouvement diffusif, la vitesse moyenne des particules est proportionnelle au gradient de potentiel
µα ∇−=rr
V . Le coefficient α est appelé mobilité; pour un système obéissant à la statistique de Boltzmann, il est relié au coefficient de diffusion Ds par la relation d'Einstein
TkD Bs=α . En appliquant cette mobilité aux adatomes de densité adρ , on obtient un courant d'adatomes
µρ
ρ ∇−==rrr
TkD
VjB
adsad .
Ceci permet de décrire l'évolution du système (voir exercice 6 sur le lissage d'une surface gravée).
Exercices 1. Soient σ l'énergie superficielle d'une surface singulière, et γ l'énergie par unité de longueur de marche sur cette surface (parfois appelée "tension de ligne"). Calculer σ pour une surface vicinale d'angle θ, en supposant que les marches sont indépendantes. Quelle est la surface d'énergie minimale?
* * * 2. On considère le modèle SOS, et on note 0σ l'énergie de surface dans la direction (001). Montrer que 2
0 6/ aHs∆=σ où a est le paramètre de maille du réseau cubique simple considéré et sH∆ est l'énergie de cohésion par atome. Calculer ensuite l'énergie d'une surface définie par ses angles par rapport à (001); tracer )(θσ . Enfin calculer la rigidité de surface.
* * * 3. On considère un solide à symétrie cubique dont l'énergie de surface est donnée pour
[ ]2,0 πθ ∈ dans le plan xy par [ ])sin()cos()( 10 θθσσθσ ++= . Tracer le diagramme polaire de σ dans ce plan. Identifier les points singuliers. Calculer la rigidité de surface en dehors des points singuliers. Calculer l'équation paramétrique de la forme d'équilibre (par exemple, calculer les coordonnées x et y du point courant M défini par θσσ uur
rr '+=OM ).

36
Commenter: courbure pour les directions non-singulières, largeur des facettes pour les directions singulières.
* * * 4. Quelle est la forme d'équilibre d'une particule de cuivre de 5mm de diamètre, mise à flotter à la surface d'un bain de plomb, dans une ampoule scellée à 1300K. On donne les énergies de surface et d'interface: surface du cuivre 1300 dyn/cm2 surface du plomb 1000 dyn/cm2 interface Cu/Pb 900 dyn/cm2 On pourra se contenter d'esquisser la solution sans développer les calculs.
* * * 5. Quelle est la forme de croissance d'un cristal cubique pour les combinaisons suivantes de vitesses de croissance: a. <001> V=0.1µm/hr <111> V=1µm/hr b. <001> V=1µm/hr <111> V=0.1µm/hr Pour quel rapport de vitesses passe-t-on d'une forme à l'autre? Quelle est l'hypothèse sur la croissance dans les autres directions?
* * * 6. Une surface est initialement lissée par recuit. On grave alors une série de traits parallèles donnant un profil sinusoïdal )/sin(),( λxAyxz = . On étudie ensuite le lissage de ce profil lors d'un nouveau recuit. Plusieurs mécanismes peuvent permettre ce lissage: diffusion de volume, diffusion de surface, évaporation-croissance,... On suppose que le mécanisme dominant est la diffusion des adatomes. On note adρ la densité d'adatome, supposée uniforme, sD leur coefficient de diffusion en surface. On admet que la vitesse moyenne des adatomes est proportionnelle au gradient de potentiel chimique à la surface du solide, le coefficient de proportionnalité (mobilité) étant TkD Bs / (relation de Nernst-Einstein). a. On suppose que la température est suffisamment haute pour utiliser le modèle continu: exprimer ),( yxµ . b. En déduire le courant j
r d'adatomes.
c. Calculer tz ∂∂ / (écrire l'équation de conservation à la surface). d. En déduire l'évolution de ),,( tyxz . On montrera que le profil reste sinusoïdal et on calculera )(tA . 7. La formule de Herring-Mullins est établie en calculant l'extremum d'une fonctionnelle (le potentiel thermodynamique), les fonctions étant les surfaces ( )yxz , , dépendant de deux variables x et y. Rapprocher ce calcul du principe de moindre action en mécanique classique, où on cherche l'extremum de l'action, fonctionnelle des trajectoires ( )tz , la variable étant le temps t.

37
IV. Hétérostructures en désaccord de maille: Contraintes et déformations élastiques
L'épitaxie est souvent réalisée entre deux matériaux de structure cristallines différentes, ou au moins dont les paramètres de maille sont différents. Si la différence est faible, on observe une épitaxie dite cohérente: le matériau déposé s'adapte à la distance entre plans atomiques du substrat, et se déforme suivant les lois de l'élasticité: c'est ce que décrit ce chapitre. Lorsque l'énergie élastique ainsi emmagasinée devient grande, on observe une relaxation des contraintes de désaccord de maille, soit par génération de défauts à l'interface (et ailleurs…), soit par un changement de la morphologie de la couche (formation d'îlots: mode de croissance Stranski-Krastanov): c'est l'objet du chapitre suivant. Plusieurs descriptions (et plusieurs jeux de paramètres) ont été introduits dans différents contextes pour décrire le lien entre une contrainte uniforme et la déformation qu'elle induit, dans le cadre de l'approximation linéaire dite élastique: - une description utilisée en résistance des matériaux, bien adaptée pour des matériaux isotropes et des contraintes simples; deux paramètres suffisent (module d'Young et coefficient de Poisson); - une description tensorielle permettant de décrire tout type de déformation ou de contrainte uniforme, au prix d'une certaine lourdeur puisqu'il faut manipuler des tenseurs d'ordre deux et quatre; - une notation abrégée issue de la description tensorielle; intermédiaire entre les deux précédentes, c'est la plus utilisée; - une description issue de la théorie des groupes, qui permet de bien prendre en compte les symétries du problème; - enfin, une description microscopique qui tient compte des liaisons chimiques dans le matériau. Les problèmes de déformation sont artificiellement compliqués par des notations parfois insuffisamment précisées, en particulier lorsqu'on passe des notations tensorielles aux notations abrégées. Je suis ici les notations de Kittel, Introduction à la physique du solide.
1. Module d'Young et coefficient de Poisson
a. Pression uniaxiale On considère un échantillon parallélipipédique (de côtés lx, ly, lz) d'un matériau isotrope et homogène, en équilibre, dont une face est soumise à une force normale F (et donc l'autre face est soumise à une force opposée). La déformation de cet échantillon ( zzz ll /∆=ε perpendiculairement à la face d'appui et xxx ll /∆=ε , yyy ll /∆=ε dans les deux autres directions) est proportionnelle à la contrainte
yxllF /=σ :
Ezσε = , où le paramètre E est appelé le module d'Young,
zx νεε −= , zy νεε −= où le paramètre ν est appelé le coefficient de Poisson.

38
Exercice: calculer l'énergie élastique emmagasinée ( zσε21 par unité de volume).
b. Cisaillement Le même échantillon est soumis à une force tangentielle sur une face et se déforme suivant un angle β. On a la relation
yxllF
G1
=β
qui définit le module de cisaillement (ou module de Coulomb) G. (Remarque: montrer que l'équilibre impose l'existence de trois autres forces tangentielles). On montre (voir 4c ci-dessous) qu'il existe une relation entre module d'Young et module de Coulomb:
)1(2 ν+= GE
c. Pression hydrostatique En combinant trois contraintes uniaxiales perpendiculaires, on obtient sans difficulté le changement de volume induit par une pression hydrostatique p.
Ep
VV
zyx )21(3 νεεε −=++=∆
2. Notation tensorielle
a. Le tenseur de déformations Une déformation uniforme d'un milieu continu homogène se décrit par le tenseur de déformation (strain): un point rr du milieu se déplace de rrδ avec ∑=
jjiji rr εδ , c'est à dire
⎟⎟⎟
⎠
⎞
⎜⎜⎜
⎝
⎛
⎟⎟⎟
⎠
⎞
⎜⎜⎜
⎝
⎛
=⎟⎟⎟
⎠
⎞
⎜⎜⎜
⎝
⎛
zyx
zyx
zzzyzx
yzyyyx
xzxyxx
εεεεεεεεε
δδδ
)( ijε est donc un tenseur d'ordre 2.
b. Le tenseur de contraintes La déformation est provoquée par une contrainte (stress), elle aussi décrite par un tenseur d'ordre 2, noté )( ijσ . Par exemple, xyσ est la composante suivant la direction x de la force exercée sur une surface élémentaire perpendiculaire à la direction y.
β

39
c. La loi de Hooke Si la réponse est linéaire, alors la déformation est liée à la contrainte par klijklklij = εσ cΣ , où
)( ijklc est un tenseur d'ordre 4, caractéristique du matériau (constantes élastiques). Ceci s'applique à des déformations faibles. On sait appliquer des contraintes telles que la déformation n'est plus linéaire: c'est en particulier le cas pour des pressions hydrostatiques, appliquées par l'intermédiaire d'un fluide dans une "enclume diamant". On doit alors considérer des termes d'ordre supérieur. Cependant le plus souvent les termes linéaires sont suffisants, la limite supérieure étant plutôt donnée par l'apparition de déformations irréversibles liées à l'apparition de défauts structuraux (le matériau devient plastique et non plus élastique). On peut aussi écrire la loi de Hooke sous la forme inverse:
klijklklij = σε sΣ
3. Les composantes de déformation et de contrainte
a. La notation abrégée A la place de ces tenseurs, on utilise souvent une notation abrégée. Le tenseur de déformation est remplacé par six composantes de déformations:
yxxy6
xzzx5
zy yz4
zz3
yy2
xx1
+ = e + = e
+ = e = e = e = e
εεεε
εεε
εε
Les termes antisymétriques sont abandonnés: ils représentent une rotation de l'ensemble du matériau. Exemple: εεεε - = , = yxxy , tous les autres étant nuls, décrit une rotation d'un angle ε autour de l'axe z. Si tout le matériau est modifié de façon uniforme, on n'a pas à tenir compte de ces termes de rotation (ce n'est plus le cas si la déformation n'est pas homogène). Attention: le fait de prendre les sommes ) + ( yxxy εε et non les demi-sommes est la source de bien des erreurs de calcul... On effectue une simplification analogue sur la contrainte, mais cette fois ce sont les demi-sommes qui sont retenues...
etc... )/2 + ( =
= = =
zyyz4
zz3yy2xx1
σσσ
σσσσσσ
Ces définitions entraînent une nouvelle définition des coefficients élastiques, qui n'ont plus que deux indices et sont rangés dans un tableau 6x6. Attention: on a simplifié la notation, mais on a masqué la nature tensorielle. Par exemple, le tenseur d'ordre quatre )(cijkl est déguisé sous forme de tableau à deux entrées )(cij .

40
b. Propriétés de symétrie Les propriétés de symétrie du matériau étudié permettent de simplifier l'écriture du tableau des constantes élastiques. Ainsi en symétrie cubique on peut montrer directement (voir Kittel) qu'il n'y a que trois termes indépendants, 441211 , et c, cc , et que l'on peut écrire:
⎟⎟⎟⎟⎟⎟⎟⎟
⎠
⎞
⎜⎜⎜⎜⎜⎜⎜⎜
⎝
⎛
⎟⎟⎟⎟⎟⎟⎟⎟
⎠
⎞
⎜⎜⎜⎜⎜⎜⎜⎜
⎝
⎛
=
⎟⎟⎟⎟⎟⎟⎟⎟
⎠
⎞
⎜⎜⎜⎜⎜⎜⎜⎜
⎝
⎛
6
5
4
3
2
1
44
44
44
111212
121112
121211
6
5
4
3
2
1
0
0
eeeeee
cc
cccccccccc
σσσσσσ
que l'on trouvera souvent écrit sous une forme hybride:
⎟⎟⎟⎟⎟⎟⎟⎟
⎠
⎞
⎜⎜⎜⎜⎜⎜⎜⎜
⎝
⎛
⎟⎟⎟⎟⎟⎟⎟⎟
⎠
⎞
⎜⎜⎜⎜⎜⎜⎜⎜
⎝
⎛
=
⎟⎟⎟⎟⎟⎟⎟⎟
⎠
⎞
⎜⎜⎜⎜⎜⎜⎜⎜
⎝
⎛
xy
zx
yz
zz
yy
xx
xy
zx
yz
zz
yy
xx
cc
cccccccccc
εεεεεε
σσσσσσ
44
44
44
111212
121112
121211
220
2
0
Ces propriétés de symétrie sont bien prises en compte par l'application de la théorie des groupes (voir le paragraphe 5 ci-dessous).
4. Quelques exemples en symétrie cubique
a. Pression hydrostatique Le matériau est dans un fluide, auquel est appliquée une pression p. Dans une enclume diamant, on peut appliquer des pressions de l'ordre de plusieurs dizaines de GPa. Chaque face du solide est soumise à la pression p, qui impose une force normale à la surface: le tenseur des contraintes a toutes ses composantes non-diagonales nulles, et ses composantes diagonales égales à -p.
ijij pδσ −= Le tenseur des déformations se calcule grâce aux coefficients élastiques: on trouve
)c 2 + /(c p- = 1211ijij δε
b. Contrainte uniaxiale Contrainte tétragonale: Considérons le cas où les deux faces opposées (001) et )100( sont soumises à une force perpendiculaire aux faces, les autres faces étant libres. L'axe de la contrainte est donc un axe d'ordre 4, d'où le terme tétragonal. L'application d'une telle

41
contrainte uniaxiale (bien définie, ce qui n'est pas si facile sur le plan expérimental), est un moyen d'étude des propriétés élastiques des matériaux, et en particulier du couplage entre les propriétés électroniques et élastiques (c'est-à-dire, par exemple, comment se déplacent les niveaux d'énergie des électrons lorsque le cristal est déformé. La seule composante non-nulle du tenseur de contrainte est pzz −=σ , où p est la force appliquée par unité de surface. On en tire sans difficulté 0 = = = zxyzxy εεε , et
pcccc
cyyxx ))(2( 12111211
12
−+== εε
pcccc
cczz ))(2( 12111211
1211
−++
−=ε
On a donc, pour un matériau cubique soumis à une contrainte uniaxiale suivant [001], un
module d'Young 1211
12111211001
))(2(cc
ccccE
zz
zz
+−+
==εσ
et un coefficient de Poisson
1211
12
ccc
zz
xx
+=−=
εε
ν .
Contrainte trigonale On peut traiter de la même façon le cas d'une contrainte uniaxiale appliquée suivant un axe d'ordre 3, c'est-à-dire sur une face (111). Le tenseur de contrainte est obtenu à partir du précédent, en effectuant une rotation qui amène l'axe de la contrainte suivant la direction [111]. La matrice de changement de base est
⎟⎟⎟⎟
⎠
⎞
⎜⎜⎜⎜
⎝
⎛
−−
3/16/203/16/12/13/16/12/1
et le tenseur de contrainte devient: ⎟⎟⎟
⎠
⎞
⎜⎜⎜
⎝
⎛−
3/3/3/3/3/3/3/3/3/
ppppppppp
.
En utilisant les symétries évidentes, on en tire sans difficulté:
)2(3 1211 ccp
zzyyxx +−=== εεε
446cpzxyzxy −=== εεε .
On en déduit le module d'Young, )2/()2(3 441211441211111 ccccccE +++= , différent du précédent. Un matériau isotrope, pour lequel contraintes trigonales et tétragonales sont équivalentes, est caractérisé par la relation 441211 2c=c-c . On verra un peu plus loin que les semiconducteurs à structure zinc-blende ou diamant, sont loin d'être isotropes: ils vérifient

42
121144 c-c>>2c , c'est-à-dire qu'ils sont beaucoup plus durs vis-à-vis d'une déformation trigonale que vis-à-vis d'une déformation tétragonale: ce comportement est lié à l'environnement tétraédrique de chaque atome, ainsi qu'on le verra un peu plus loin en décrivant les modèles microscopiques de la déformation.
c. Cisaillement Le cas simple est celui d'un solide en équilibre, sur lequel on applique une contrainte 0≠xyσ ; l'équilibre impose xyyx σσ = (le moment appliqué est nul). Les autres composantes sont nulles. En passant au tenseur de déformation on obtient 442/ cxyxyxy σεε == . On peut remarquer sur la figure que l'angle 6eyxxy =+= εεβ (ce qui justifie cette définition de 6e ), d'où 44cG = . Pour un matériau isotrope, on a donc )1(2 ν+= GE .
Fig.1: cisaillement d'un objet carré en équilibre (à gauche) identifiant l'angle de déformation (au milieu) et le tenseur de déformation (à droite)
d. Déformation dans une couche épitaxiée Lorsqu'un matériau est épitaxié sur un substrat, il y a continuité du réseau cristallin à travers l'interface entre les deux matériaux. Cependant, on peut aussi avoir épitaxie lorsque les deux matériaux ont des paramètres de maille légèrement différents. Dans ce cas, la couche va subir une déformation dans le plan de croissance, de manière à ajuster sa maille cristalline dans ce plan à celle du substrat. L'hypothèse la plus simple est celle d'une déformation uniforme de la couche (voir figure); dans ce cas, si l'on suppose que le substrat est infiniment plus épais que la couche déposée, seule la couche est déformée. Cette déformation se décrit à l'aide du formalisme de l'élasticité. On va traiter l'exemple le plus simple: un matériau à structure cubique, de paramètre de maille a, est déposé sur une face (001) d'un autre matériau de structure cubique, de paramètre sa . On choisit comme habituellement x // [100], y // [010], z // [001]. La déformation est homogène dans toute la couche. Le substrat n'est pratiquement pas déformé (en fait on a une courbure de l'échantillon, mais cet effet est négligeable au niveau microscopique). Les conditions aux limites sont: - la déformation dans le plan de la couche est telle que le réseau cristallin de la couche est identique à celui du substrat:
0 =
a)/a-(a = =
xy
syyxx
ε
εε
.
- la surface est libre, donc les contraintes y sont nulles: 0 = = = zyzxzz σσσ .
β εxy
εyx

43
Ces six conditions aux limites, et les relations contrainte/déformation, permettent de calculer les autres composantes de déformation:
aa-a
c2c- =
0 = =
s
11
12zz
zyzx
ε
εε
Exercice: écrire ces deux formules en utilisant le module d'Young et le coefficient de Poisson tétragonaux, et montrer que la densité d'énergie élastique dans la couche s'écrit
2
1⎟⎠⎞
⎜⎝⎛
−=
aaEEel
δν
.
Cette expression est valable pour un matériau isotrope quelle que soit l'orientation de la croissance épitaxiale. Pour un matériau cubique, elle n'est valable qu'en orientation (001) ou (111), avec les coefficient de Poisson et module d'Young adaptés.
5. La prise en compte des symétries Dans le paragraphe précédent, on a calculé les tenseurs de déformation et de contrainte d'un matériau à symétrie cubique, dans quelques cas simples où la contrainte et la déformation se font suivant des axes de haute symétrie. Pour une contrainte ou une déformation quelconque (exemple: couche épitaxiée sur une surface d'orientation différente de (001) ou (111)), le calcul devient rapidement plus difficile. Pour bien comprendre le rôle des symétries, on utilise les résultats de la théorie des groupes. Celle-ci permet de classer les tenseurs d'ordre 2 d'un matériau à symétrie cubique suivant quatre types, chaque type étant associé à une "représentation" du groupe de symétrie.
totalement symétrique: ⎟⎟⎟
⎠
⎞
⎜⎜⎜
⎝
⎛=Γ
100010001
)( 1M
tétragonal: ⎟⎟⎟
⎠
⎞
⎜⎜⎜
⎝
⎛−
−=
200010001
31)(θM et
⎟⎟⎟
⎠
⎞
⎜⎜⎜
⎝
⎛−=
000010001
)(εM
Fig. 2: Déformation d'un matériau A à grand paramètre de maille en épitaxie sur un matériau B à paramètre de maille plus petit. La maille cubique du matériau A (à gauche, hors épitaxie) est comprimée dans le plan de la couche et s'allonge suivant l'axe de croissance (à droite).

44
trigonal: ⎟⎟⎟
⎠
⎞
⎜⎜⎜
⎝
⎛=
010100000
)(ξM , ⎟⎟⎟
⎠
⎞
⎜⎜⎜
⎝
⎛=
001000100
)(ηM , et ⎟⎟⎟
⎠
⎞
⎜⎜⎜
⎝
⎛=
000001010
)(ζM
antisymétrique: ⎟⎟⎟
⎠
⎞
⎜⎜⎜
⎝
⎛
−=
010100000
)(xM , ⎟⎟⎟
⎠
⎞
⎜⎜⎜
⎝
⎛ −=
001000100
)(yM , et ⎟⎟⎟
⎠
⎞
⎜⎜⎜
⎝
⎛−=
000001010
)(zM
Un tenseur de déformations proportionnel à l'un de ces tenseurs correspondra à: - la déformation totalement symétrique (dilatation, positive ou négative), en réponse à une pression hydrostatique. - un cisaillement tétragonal, dont l'axe est l'un des axes principaux du cube, [100], [010] ou [001]. - un cisaillement trigonal, dont l'axe est un axe <111>. On définit ainsi six déformations élémentaires, et toute déformation peut être écrite sous forme de combinaison linéaire de ces déformations élémentaires (en effet, si l'on exclut les trois termes antisymétriques du tenseur qui représentent une simple rotation de l'échantillon, il nous reste six composantes indépendantes). Ces déformations élémentaires sont schématisées sur la figure:
Pourquoi avoir regroupé ces tenseurs en "représentations"? Ce classement est lié au comportement des tenseurs élémentaires ainsi définis lors des opérations de symétrie du groupe du cube: ces tenseurs se transforment entre eux à l'intérieur d'une représentation. Ainsi, considérons une symétrie par rapport au plan (011), ou plan yz. Cette opération échange l'axe y et l'axe z, l'axe x restant invariant. On obtient sans peine que: - le tenseur totalement symétrique est invariant (représentation 1Γ ) - les tenseurs de type tétragonal se transforment suivant:
⎥⎦
⎤⎢⎣
⎡×⎥
⎦
⎤⎢⎣
⎡
−−−
→⎥⎦
⎤⎢⎣
⎡)()(
2/12/32/32/1
)()(
εθ
εθ
MM
MM
Ce comportement est celui qui est défini dans les tables de théorie des groupes comme caractéristique de la représentation 3Γ .
Fig. 3: Les déformations élémentaires (Γ1, Γ3, Γ5 ), et leur effet en symétrie sphérique (expansion et cisaillement) ou en symétrie cubique (expansion, cisaillement tétragonal et cisaillement trigonal)

45
- parmi les tenseurs de type trigonal, )(ξM est invariant alors que )(ηM et )(ζM s'échangent. Ce comportement est caractéristique de la représentation 5Γ . On pourra écrire des transformations analogues pour les autres opérations de symétrie.
L'intérêt de tels regroupement est que si une relation est établie entre deux tenseurs élémentaires, la même relation (avec les mêmes paramètres) existera entre tous les tenseurs appartenant à la même représentation. On va maintenant en voir l'illustration dans le cas de l'élasticité.
b Application aux exemples précédents. Pression hydrostatique: On a
)()( 1Γ−= Mpijσ et )( 2
1- = )( 11211
ij ΓMp c + c
ε
A une pression hydrostatique est donc associée une déformation totalement symétrique. La combinaison des coefficients élastique ) c + (c 1211 2 est ainsi liée à la représentation 1Γ du groupe du cube. Contrainte uniaxiale. Pour une contrainte uniaxiale suivant [001)], le calcul précédent se met sous la forme
( ) )(3
)(3 1 θσ MM pp
ij−
+Γ−
=
( ) )(3
1)(32
1
12111
1211
θε MM pcc
pccij
−−
+Γ−
+=
Une contrainte uniaxiale suivant un axe <100> fait donc apparaître une composante de déformation tétragonale et une composante hydrostatique. Liée à la composante hydrostatique, on retrouve la combinaison de constante élastique )c+(c 1211 2 . Liée au cisaillement tétragonal, on trouve )( 1211-cc . Pour une contrainte uniaxiale suivant [111], on a obtenu
( ) [ ])()()(3
)(3 1 ζηξσ MMMM ++
−+Γ
−=
ppij
( ) [ ])()()(32
1)(32
1
441
1211
ζηξε MMMM ++−
+Γ−
+=
pc
pccij
Une contrainte uniaxiale suivant l'axe [111] induit donc une déformation hydrostatique, pour laquelle on retrouve bien la combinaison ) c+c 1211 2( , et un cisaillement trigonal avec le coefficient élastique 442c . Cisaillement On a bien trouvé le paramètre 442c entre le cisaillement )()( ζεε M=ij et la contrainte
)()( ζσσ M=ij .

46
Déformation dans une couche épitaxiée. Pour une couche épitaxiée sur un substrat d'orientation (001), on a obtenu
)(23
1)(32)(
11
12111
11
1211 θε MMc
ccc
ccij
+−Γ
−=
( ) ( )⎥⎦
⎤⎢⎣
⎡−Γ
+−= )(
31)(
322)( 1
11
12111211 θδσ MMaa
ccccc
ij
où on retrouve bien une composante hydrostatique avec le rapport ( )1211 2cc + et une composante tétragonale avec le rapport ( )1211 cc − . On peut écrire une expression analogue, avec une composante hydrostatique et une composante trigonale, dans le cas d'une couche épitaxiée sur un substrat d'orientation (111). Pour plus de détails sur les propriétés de symétrie, un "classique": N.F.Nye, Physical properties of crystals: their representation by tensors ans matrices, Oxford, Clarendon Press
6. Modèles microscopiques. Le tenseur de déformation a été construit pour décrire un milieu continu. Pour un milieu cristallin, il permet de décrire non seulement une déformation macroscopique, mais aussi la déformation de la maille élémentaire. Cependant, pour un semiconducteur: - il ne décrit pas ce qui se passe à l'intérieur de la maille élémentaire, c'est à dire le mouvement relatif des deux atomes de la maille (ou des deux sous-réseaux). - il ne tient compte que de la symétrie cubique, et pas de l'existence du tétraèdre local qui définit les directions de liaisons. Pourtant lorsque on déforme un cristal, ce sont bien les liaisons qui vont opposer une résistance. Les modèles les plus simples qui répondent à ces objections ont été adaptés de la physique moléculaire: ils sont dénommés Valence Force Field. On va décrire ici le modèle donné par Harrison, (W.A.Harrisson, Electronic structures and the properties of solids, Freeman, San Francisco, 1981). Un modèle proche, plus facile à mettre en œuvre dans les calculs numériques et donc très utilisé, mais moins intuitif, est celui de Keating (Phys. Rev. 145, 637 (1966)). Ils tiennent compte de deux types de déformations au niveau local: - changement de longueur des liaisons (bond stretching). L'énergie est écrite 2
0 d/d)( C 1/2 δ , où d est la longueur de la liaison. Il y a quatre liaisons par paire d'atomes. - changement de l'angle entre deux liaisons du même atome (bond bending). L'énergie est
( )212
1 δθC , où θ est l'angle entre deux liaisons (par exemple Zn-S-Zn).Il y a six angles par atome, douze par paire. Ces deux paramètres, 0C et 1C , doivent rendre compte des trois constantes élastiques, 11c ,
12c , et 44c , mais aussi décrire la déformation interne de la maille. Ils ne s'en tirent pas trop mal, à 10 ou 15 % près. En pratique 0C et 1C sont calculés en considérant une déformation hydrostatique (qui ne joue que sur les longueurs de liaisons) et une déformation tétragonale (dont on montre qu'elle ne joue que sur les angles: figure 13). On obtient (voir, par exemple, Harrison, 1981, pour la démonstration; on pourra aussi la faire soi-même!):

47
( )1211
30
1
1211
30
0
32
)2(163
cca
C
cca
C
−=
+=
Par ailleurs, une déformation trigonale modifie à la fois les angles et les longueurs de liaisons, et fait donc intervenir 0C et 1C . Le calcul est plus compliqué (voir Harrisson).
Fig. 4: Rotation des liaisons dans une déformation de cisaillement tétragonal de type Γ3ε. Le tétraèdre est vu suivant l'axe z = [001].
Exercices On dépose, en épitaxie cohérente, une couche fine (3 monocouches) de ZnTe sur un substrat CdTe d'orientation (100). Calculer les tenseurs de déformation et de contrainte, et l'énergie élastique. Comparer la composante totalement symétrique à l'effet de la pression atmosphérique usuelle d'une part, à la pression hydrostatique de 20 GPa appliquée en laboratoire dans une enclume diamant d'autre part. paramètre de maille de CdTe: a0=6.48Å paramètre de maille de ZnTe: a0=6.10Å Coefficients élastiques de ZnTe: c11=7.13×1010 Pa c12=4.07×1010 Pa c44=3.12×1010 Pa Même question en orientation (111). Commentez. Que pensez-vous de l’éventualité de considérer ZnTe comme un solide isotrope?
x=[100]
y=[010]

48

49
V. Relaxation des contraintes: dislocations et îlots On a vu dans le chapitre d'introduction que, lorsque l'épaisseur d'une couche épitaxiée croît, des défauts apparaissent à l'interface et dans la couche. Les défauts à l'interface sont des dislocations de désaccord de maille (misfit dislocations): elles relaxent une partie du désaccord de maille et donc de la contrainte. Les défauts dans la couche sont des "threading dislocations" et sont dûs au mécanisme de formation des "misfit dislocations". L'apparition de ces défauts dégrade les propriétés électroniques du matériau, en particulier dans le cas des semiconducteurs (les métaux sont moins sensibles).
1. Dislocations Les dislocations sont (après les défauts ponctuels: lacune, interstitiel, …) les plus simples des défauts cristallins. Elles interviennent dans les propriétés de plasticité, dans les joints de grains… et la relaxation des couches contraintes. Pour une description simple, voir Kittel, Introduction à la Physique du solide; pour des descriptions plus complètes, voir J.Friedel Dislocations (Pergamon 1964) ou Hirth et Loth, Theory of Dislocations, (Wiley 1982). On se contentera ici d'une description très simple. La définition générale est la suivante. On considère un circuit L dans le matériau ("ligne de dislocation"), on déplace d'un vecteur b
r("vecteur
de Burgers") le matériau situé d'un côté d'une surface s'appuyant sur L, enfin on complète le matériau manquant ou on enlève l'excès. Deux remarques immédiates: - Pour que la dislocation soit compatible avec la structure cristalline, le vecteur de Burgers doit être
Fig. 1: Images (vue en plan) d'une couche GaInAsP réalisées suivant différents modes du microscope électronique: (A) Electron Beam Induced Current, (B) cathodoluminescence; en bas, images en mode classique du rectangle indiqué en haut (Petroff et al, Phys. Rev. Lett. 44 (1980) 287). En bas, différents types de dislocations sont mis en évidence en utilisant différents vecteurs de diffraction des électrons. En haut, les lignes noires montrent l'effet de ces dislocations sur les électrons dans le semiconducteur: les électrons injectés par le microscope donnent lieu à un courant (EBIC) ou à une émission de photons (CL), sauf lorsqu'ils sont injectés à proximité des dislocations.
Fig. 2: Schéma général d'une dislocation (Friedel, Dislocations)

50
un vecteur du réseau cristallin. - La ligne est fermée, ou intercepte la surface de l'échantillon ("dislocation émergente"). Si on décompose la ligne en segments, toute dislocation peut être considérée comme une combinaison de: - dislocations vis: Lb
rr//
- dislocations coin: Lbrr
⊥
A partir de là, on peut former des dislocations plus compliquées avec une nomenclature particulière à chaque type de structure cristalline (exemple fig.5). Enfin, une fois qu'une dislocation est repérée sur une image de microscopie à haute résolution, le vecteur de Burgers est déterminé en utilisant un circuit de Burgers: on effectue d'une part un circuit fermé , en sautant d'un atome à l'autre, autour de la dislocation, et d'autre part un circuit défini par les mêmes sauts, dans une zone sans défaut. Le second circuit ne se referme pas, et la distance entre les deux extrémités est le vecteur de Burgers.
Fig. 3: Dislocation coin (a) et vis (b) (Friedel, Dislocations)
Fig. 4: La plus simple des dislocations coin: dans un cristal cubique (Kittel)
Fig. 5: Dislocation dite "de Lomer" en structure diamant (F.Louchet et J Thibault-Desseaux, Rev. Phys. Appl. 22 (1987) 207). Suggestion: tracer le circuit de Burgers sur l'image répétée à droite.

51
Des expressions détaillées existent pour le champ de déformation et l'énergie des différents types de dislocations. Le calcul est très simple dans le cas d'une dislocation vis: le matériau décrit une hélice autour de la ligne, de pas b. A une distance ρ de la ligne, on a donc un cisaillement d'angle ρπε 2/b= et la contrainte est ρπεσ 2/GbG == . L'énergie s'obtient
en intégrant ⎟⎠⎞
⎜⎝⎛== ∫ r
RbGLdLER
rdisl Ln
42 2
21
πρπρσε . On a une énergie par longueur de ligne
L. Perpendiculairement à la ligne, le volume est limité à grande distance par tout objet limitant l'extension du champ de déformation: dislocation voisine, surface de l'échantillon, etc… C'est ce qui fixe la valeur de R. A courte distance, la coupure r (de l'ordre du paramètre de maille) est un fourre-tout qui tient compte de la structure cristalline (limite de validité de la théorie continue de l'élasticité, mais aussi autres contributions à l'énergie: liaisons coupées, reconstruction du cœur de la dislocation, …). Pour une dislocation coin, le calcul est plus compliqué, mais le résultat très proche:
( ) ⎟⎠⎞
⎜⎝⎛
−=
rRbG
LEdisl Ln
142
νπ
Enfin, les dislocations bougent (c'est à dire que la ligne se déplace, perpendiculairement à L
r),
mais leur mouvement est très anisotrope: - le mouvement parallèlement à b
r, dit de glissement, est très facile; il correspond à une
réorganisation des liaisons au cœur de la dislocation; - le mouvement perpendiculairement (la dislocation "grimpe") implique la capture ou l'émission de défauts ponctuels (lacunes, interstitiels) qui permettent d'étendre ou de rétrécir le circuit L, et il est plus difficile et activé thermiquement.
2. Dislocations de misfit et épaisseur critique
L'hypothèse la plus simple est celle d'un réseau de dislocations coins, de vecteur de Burgers parallèle à l'interface, dont les lignes sont à l'interface, dans deux directions perpendiculaires. Si n est la densité linéaire de ce réseau de dislocations (c'est-à-dire que la distance moyenne entre deux dislocations est 1/n), la déformation
dans la couche ("déformation résiduelle") devient nbaa−=
δε et la densité d'énergie élastique
est diminuée: 2
1⎟⎠⎞
⎜⎝⎛ −
−= nb
aaEEelδ
ν.
L'énergie totale, par unité de surface, est donc (en supposant que c'est la distance à la surface qui limite l'extension du champ de contrainte des dislocations):
( ) ⎟⎠⎞
⎜⎝⎛
−+⎟
⎠⎞
⎜⎝⎛ −
− rhbGnhnb
aaE Ln
142
12
2
νπδ
ν.
Sur une longueur L d'interface, on a L/as rangées atomiques arrivant du côté substrat. Du côté couche, aux L/as rangées atomiques qui traversent l'interface s'ajoutent nL rangées qui se terminent par une dislocation; en l'absence de contrainte, le tout occuperait une longueur (Lac/as+nLb)=L(1-δa/a+nb). D'où la valeur de la déformation.
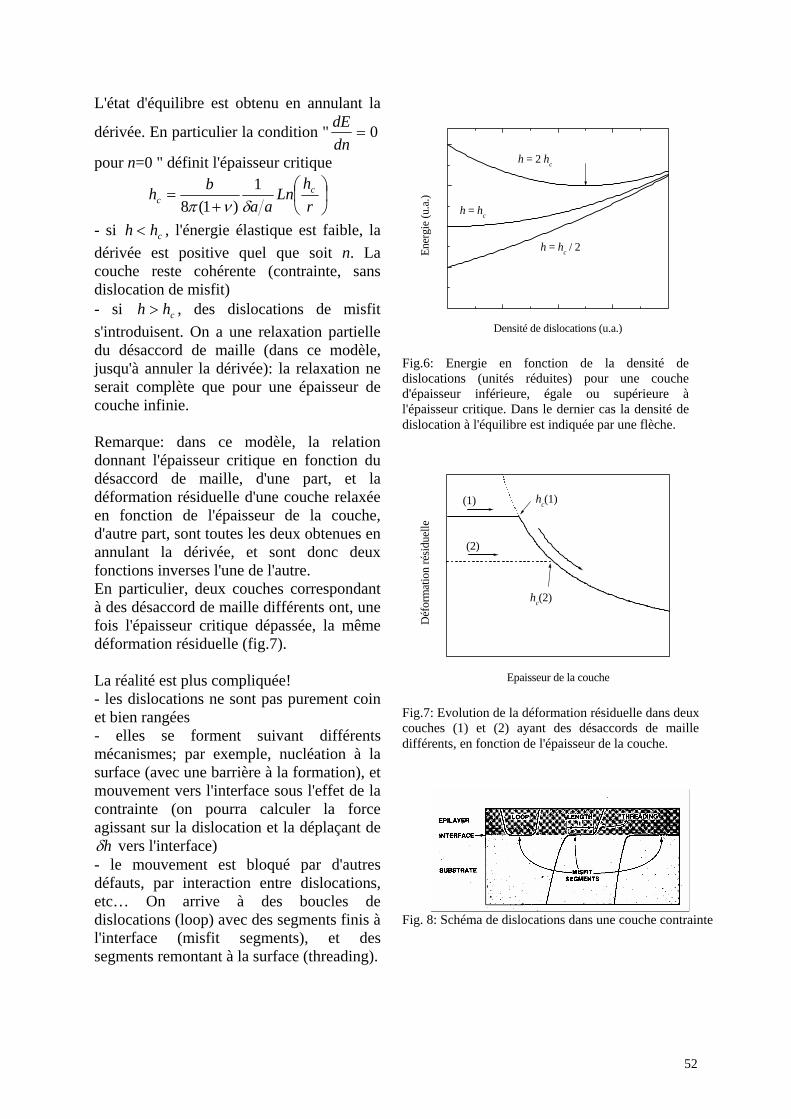
52
L'état d'équilibre est obtenu en annulant la
dérivée. En particulier la condition " 0=dndE
pour n=0 " définit l'épaisseur critique
⎟⎠⎞
⎜⎝⎛
+=
rh
Lnaa
bh cc δνπ
1)1(8
- si chh < , l'énergie élastique est faible, la dérivée est positive quel que soit n. La couche reste cohérente (contrainte, sans dislocation de misfit) - si chh > , des dislocations de misfit s'introduisent. On a une relaxation partielle du désaccord de maille (dans ce modèle, jusqu'à annuler la dérivée): la relaxation ne serait complète que pour une épaisseur de couche infinie. Remarque: dans ce modèle, la relation donnant l'épaisseur critique en fonction du désaccord de maille, d'une part, et la déformation résiduelle d'une couche relaxée en fonction de l'épaisseur de la couche, d'autre part, sont toutes les deux obtenues en annulant la dérivée, et sont donc deux fonctions inverses l'une de l'autre. En particulier, deux couches correspondant à des désaccord de maille différents ont, une fois l'épaisseur critique dépassée, la même déformation résiduelle (fig.7). La réalité est plus compliquée! - les dislocations ne sont pas purement coin et bien rangées - elles se forment suivant différents mécanismes; par exemple, nucléation à la surface (avec une barrière à la formation), et mouvement vers l'interface sous l'effet de la contrainte (on pourra calculer la force agissant sur la dislocation et la déplaçant de
hδ vers l'interface) - le mouvement est bloqué par d'autres défauts, par interaction entre dislocations, etc… On arrive à des boucles de dislocations (loop) avec des segments finis à l'interface (misfit segments), et des segments remontant à la surface (threading).
Ener
gie
(u.a
.)
Densité de dislocations (u.a.)
h = hc
h = 2 hc
h = hc / 2
Fig.6: Energie en fonction de la densité de dislocations (unités réduites) pour une couche d'épaisseur inférieure, égale ou supérieure à l'épaisseur critique. Dans le dernier cas la densité de dislocation à l'équilibre est indiquée par une flèche.
hc(2)
hc(1)
Déf
orm
atio
n ré
sidue
lle
Epaisseur de la couche
(2)
(1)
Fig.7: Evolution de la déformation résiduelle dans deux couches (1) et (2) ayant des désaccords de maille différents, en fonction de l'épaisseur de la couche.
Fig. 8: Schéma de dislocations dans une couche contrainte

53
3. Ilots tridimensionnels; croissance Stranski-Krastanov
a. Déformation dans un îlot On considère toujours une couche épitaxiée mouillante, contrainte; le début de croissance se fait monocouche par monocouche, et l'énergie élastique dans une telle couche d'épaisseur h
uniforme est hLaaEEel
22
1⎟⎠⎞
⎜⎝⎛
−=
δν
(où L est la largeur de couche considérée). Le substrat
épais n'est pas déformé. Si des îlots se forment, la contrainte se relaxe partiellement: chaque monocouche sera d'autant moins contrainte qu'elle sera plus loin de l'interface. Le calcul passe par la résolution d'une équation différentielle (voir un traité sur l'élasticité comme Landau et Lifshitz), dont la résolution conduit à remplacer le tenseur élastique ijklc (qui relie une déformation uniforme à une contrainte uniforme) par une fonction qui relie la déformation locale au point rr à la contrainte locale en un autre point 'rr . Cependant, on peut comprendre intuitivement ce qui se passe en utilisant le Principe de Saint-Venant: suivant celui-ci, lorsqu'une contrainte est appliquée localement (sur un volume de dimension L ou avec une période Λ), elle induit une déformation qui s'étend sur une longueur de l'ordre de L (ou Λ). Ainsi un îlot de dimensions
hLL ×× avec h>>L sera contraint au voisinage du substrat sur une hauteur de l'ordre de h, et complètement relaxé à son sommet (fig.9b). On peut écrire l'énergie élastique résiduelle sous la forme:
⎟⎠⎞
⎜⎝⎛
⎟⎠⎞
⎜⎝⎛
−=
LhRhL
aaEEel
22
1δ
ν,
où la fonction de relaxation vaut 1 pour h/L<<1 et hL /≈ pour h/L>>1. Cette fonction, peut être calculée quasi analytiquement pour des îlots de forme simple (Müller et Kern, J. Crystal Growth 193 (1998) 257; Surface Sci. 392 (1997) 103); elle est le plus souvent calculée numériquement; elle doit aussi tenir compte d'effet supplémentaires, comme la déformation du substrat (qui est maintenant soumis à une contrainte inhomogène) et les effets de bords d'îlots.
La formation d'îlots diminue l'énergie élastique, mais elle augmente la surface exposée. On note tout de suite qu'il va rester une "couche de mouillage" d'au moins une monocouche: puisque sid σσσ <+ (condition de mouillage initial), exposer le substrat augmenterait
Fig. 9: Schéma des déformations pour des îlots parallélépipédiques (Bimberg et al, Quantum Dot Heterostructures, Wiley 1999)
Fig. 10: Relaxation dans des îlots pyramidaux, en fonction de l'angle entre la facette et le substrat (ibid.)

54
l'énergie de surface. Pour des îlots parallélépipédiques, on ajoute l'énergie des flancs, Lhσ4 . L'énergie totale est donc
⎟⎠⎞
⎜⎝⎛
⎟⎠⎞
⎜⎝⎛
−+⎟
⎠⎞
⎜⎝⎛
LhRV
aaE
LhV
231
3/2
14 δ
νσ
où V est le volume de l'îlot et h/L caractérise sa forme. La forme optimale (rapport d'aspect h/L) éventuelle de l'îlot est obtenue en dérivant cette expression par rapport à h/L.
b Croissance Stranski-Krastanov Le modèle précédent est très fruste. Dans la réalité, ce mode de croissance, dit de Stranski-Krastanov, se déroule comme suit (Müller et Kern, Appl. Surf. Sci 102 (1996) 6): - au début, dépôt monocouche par monocouche, c'est à dire qu'entre deux monocouches complètes on a des îlots 2D (d'une monocouche d'épaisseur) - au dessus d'une épaisseur déposée hc, les îlots 2D suffisamment gros se transforment en îlots 3D, épais. Ceci se fait aux dépens du matériau déjà déposé. - il reste une couche de mouillage, d'épaisseur inférieure à hc . Et bien sûr la dynamique intervient aussi: si la diffusion en surface est trop faible, les îlots ne pourront pas se former, ou la couche de mouillage restera plus épaisse. On a donc un nouveau mode de relaxation des contraintes, par formation d'îlots, qui entre en compétition avec la relaxation par dislocations de misfit. L'un fait intervenir énergie élastique et énergies de surface et d'interface (et diffusion de surface), l'autre énergie élastique et énergies de dislocations 'et barrières à leur formation). On pourra donc favoriser l'un ou l'autre des deux mécanismes par le choix du matériau ou des conditions de croissance. La croissance en îlots permet de réaliser des structures à confinement dans les trois directions (cas des semiconducteurs), ou des structures où l'énergie électrostatique devient forte (blocage de Coulomb, électronique à un électron des métaux). Il "suffit" de former les îlots à la surface de l'échantillon en cours de croissance… puis de refermer le tout par une couche uniforme. Ce mode d'élaboration est souvent opposé aux méthodes utilisant la gravure, qui introduisent des défauts aux interfaces. Le contrôle précis de cette "croissance auto-assemblée" reste cependant difficile. En effet la forme finale résulte de plusieurs processus mal maîtrisés: nucléation d'un îlot, dynamique de croissance de l'îlot à partir des adatomes voisins, mûrissement partiel vers une forme d'équilibre, modification des îlots (dilution, diffusion) lorsque la couche de couverture est déposée. Les fluctuations de taille et de forme restent fortes (typiquement 20% dans les bons cas, voir la seconde partie du cours).
c. Croissance auto-organisée La source principale de fluctuation est le caractère aléatoire de la nucléation des îlots. On cherche donc des méthodes pour piloter cette nucléation par une inhomogénéité régulière de la surface. Plusieurs mécanismes peuvent jouer un rôle: - reconstruction de surface - réseau de marches d'un substrat désorienté - contraintes liées à un réseau de dislocations enterré (figure 11)

55
- contraintes liées à des îlots enterrés: la contrainte à la surface de l'échantillon où des îlots ont été enterrés agit pour favoriser ou défavoriser la nucléation, et on a un effet de tri d'une couche d'îlots à la suivante (figure 12); voir aussi la figure 9 du chapitre d'introduction.
Fig. 11: En haut: a/ image STM avec un réseau de dislocations après dépôt de 2 monocouches Ag sur Pt(111); b/ îlots de Fe déposés sur la surface précédente; c/ distribution des tailles d'îlots lors du dépôt sur réseau de dislocations ou directement sur Pt; d/ détail des îlots; e/ même dépôt, sur une partie de l'échantillon recouvert de 2 monocouches ( le réseau de dislocation est formé et les îlots s'organisent) ou 1.5 monocouches (pas de dislocations, nucléation aléatoire) Brune et al., Nature 394, 451 (1998)
Fig. 12: Ilots PbSe sur PbTe (111): A/ Image AFM des îlots: B/ une seule couche de PbSe sur PbEuTe: distribution aléatoire d'îlots (cf la transformée de Fourier en insert); C/ la soixantième couche d'îlots: distribution ordonnée. Springholz et al, Science 282, 734 (1999)
e

56

57
Seconde partie
Propriétés électroniques
Mise en évidence du confinement latéral lors de la transmission d'un électron à travers un nanocontact
Topinka et al., Science 289, 2323 (2000)

58

59
I. Confinement Le but de ce chapitre est de décrire les effets de confinement qui apparaissent lorsque le matériau est structuré dans une, deux ou les trois directions à une échelle de l'ordre de 1/k, où k est le vecteur d'onde des électrons étudiés. Ces échelles sont plus faciles à atteindre dans les semiconducteurs ( ≥Fk/1 10 nm) que dans les métaux ( ≈Fk/1 fraction de nm). Ces effets de confinement sont mis en évidence directement dans des mesures de spectroscopie (ils sont même utilisés dans des composants comme les lasers à semiconducteurs). Leur compréhension constitue également un préalable à l'étude des propriétés de transport (prochain chapitre). Dans ce chapitre, on considère d'abord le cas d'un "seul" électron dans un puits quantique ou une boîte quantique, puis les interactions entre plusieurs électrons confinés dans une boîte.
1. Systèmes 2D : confinement dans une direction Problème élémentaire de la mécanique quantique : on considère une particule de masse m, dans un potentiel à une dimension V(z). On oublie les autres directions. On doit résoudre une équation de Schrödinger
)()()()(2 2
22
zEzzVzzm
ϕϕϕ =+∂∂
−h .
Réalisation pratique : Une fine couche de semiconducteur à petit gap dans un semiconducteur de gap plus grand (GaAs dans Ga1-xAlxAs, ou GaN dans AlN, ou CdTe dans Cd1-xMgxTe, etc...). Dans le cas de la bande de conduction d'un semiconducteur à gap direct, on justifie bien l’approximation de la fonction enveloppe, qui est décrite en appendice à ce chapitre. L'essentiel de la méthode peut se résumer comme suit. La fonction d’onde d’un électron dans la bande de conduction du semiconducteur infini, parfait, uniforme, est une fonction de Bloch, ).exp()()( rkirur kk
rrrrrv =ψ . Le vecteur d'onde k
rest
dans la première zone de Brillouin, et la fonction )(ruk
rr est périodique. Le cas le plus simple
est celui d'un semiconducteur dont la bande de conduction a son minimum, E0, au centre de la zone de Brillouin k=0. Au voisinage de k=0, la dépendance en k de l'énergie se trouve être
isotrope, et peut être décrite par un développement limité au deuxième ordre, *22
0 2/ mkEEk h+= . Ceci définit la "masse effective" m*, qui ne représente à ce stade que la courbure de la fonction E(k). La méthode de la fonction enveloppe va exploiter la similitude de cette expression avec l'énergie cinétique d'une particule. Pour décrire l'effet d'un potentiel supplémentaire )(rV r , on recherche la fonction d’onde sous la forme (fig. 1)
)()()( 0 rFrur rrr=ψ , et on montre qu'on a
à résoudre une équation différentielle qui porte sur la fonction enveloppe )(rF r et
Fig. 1 : Partie périodique de la fonction de Bloch (en haut: la période est définie par la maille élémentaire), et fonction d'onde localisée par un potentiel (en bas, avec une fonction enveloppe en pointillés)

60
s'écrit
)()()()()(2 0*
2
rFEErFrVrFm
rrrrh−=+∆− .
On va appliquer la méthode à un système pour lequel elle n'était pas prévue au départ: un empilement de couches de matériaux semiconducteurs différents, sans potentiel de perturbation V explicite. On peut écrire l'équation différentielle dans chacun des matériaux:; en supposant que la masse effective est la même dans les deux matériaux (cette hypothèse n'est pas indispensable mais simplifie les calculs), la seule différence est la valeur de l'énergie au bas de la bande de conduction, qu'on notera BE0 dans le matériau où elle est plus grande (matériau "barrière") et 0E dans le matériau où elle est plus faible (matériau "puits"). On a donc deux équations différentielles,
)()()(2 0*
2
rFEErFm
B rrh−=∆− dans le matériau barrière, et
)()()(2 0*
2
rFEErFm
rrh−=∆− dans le matériau puits.
On peut rassembler ces deux équations en une seule, si on définit un potentiel 0)( =rV r dans le matériau puits et 000)( EEVrV B −==
r dans le matériau barrière.
Dans le cas d'un puits quantique constitué d'une seule couche de matériau "puits" séparée du matériau "barrière" par une interface parfaite, le potentiel ne dépend que de la coordonnée z normale à l'interface:
0)()( VzVrV ==r
en si 2/Lz > , et 0 dans le
puits 2/Lz < . Puisque le potentiel est invariant en x et y, le problème est séparable. Plus précisément, les
opérateurs 2
2
*
2
2 xm ∂∂
−h , 2
2
*
2
2 ym ∂∂
−h et
( )zVxm
+∂∂
− 2
2
*
2
2h commutent dans l'espace des états à trois dimensions (fonctions à trois
variables x, y, z): on peut donc trouver une base propre commune à cet ensemble complet d'observables qui commutent. Le premier opère sur la seule variable x, ses vecteurs propres dans l'espace des fonctions de la variable x sont les ondes planes ( )xik xexp et elles forment une base de cet espace. Il en va de même pour le deuxième, avec les ondes planes ( )yik yexp . Pour le troisième, il faut résoudre l'équation
)()(2
)()()(2
22*
2
02
2
*
2
zFkkm
EEzFzVzFzm yx ⎟⎟
⎠
⎞⎜⎜⎝
⎛+−−=+
∂∂
−hh ,
et les solutions forment encore une base de l'espace des fonctions de la variable z. Finalement, les produits de ces trois types de fonctions, ( ))(exp)()( ykxkizFrF yx +=
r , forment une base des états dans l'espace à trois dimensions (fonctions des trois variables), base dont les vecteurs sont des états propres de chacun des trois hamiltoniens qui commutent entre eux,
zLz/2-Lz/2
E-E0
0
E0B-E0
Puits quantique : profil du potentiel

61
mais également de leur somme qui est l'hamiltonien total. On a donc trouvé une base propre de l'hamiltonien total. On a donc séparé les variables et l’équation qui reste à résoudre est celle des chapitres d’introduction à la mécanique quantique. Mais il ne faut pas oublier les deux degrés de libertés qui restent (x et y, parallèles au couches), qui font qualifier ces systèmes de "bidimensionnels" ou "2D". Cette approximation que constitue l'utilisation de la méthode de la fonction enveloppe est bien justifiée (voir le cours de Physique des Semiconducteurs) si l'on étudie le confinement d'un électron de la bande de conduction dans un puits quantique formé d'un semiconducteur à gap direct (c'est-à-dire dont le minimum de la bande de conduction est en k=0), et si la fonction enveloppe F(z) qu'on trouve varie lentement (typiquement quelques mailles élémentaires). Bien que plus difficile à justifier ou même parfois non justifiée, elle sera utilisée pour une approche simple des autres cas : bande de conduction ayant un minimum au voisinage de k non nul (exemple: silicium), trou dans la bande de valence, autres matériaux….
a. Puits infini : rappels On suppose que la hauteur des barrière est grande (V0 ∞→ ). Alors la fonction d'onde est nulle dans les barrières et on obtient facilement les états propres confinés, indexés par un entier n. Système modèle : Dans le puits, on a un hamiltonien dont les solutions sont des ondes planes; deux ondes planes en k et –k ont la même énergie *22 2mkh et on doit prendre une combinaison linéaire qui s'annule aux interfaces. Compte tenu de la symétrie du problème, il s'agit des combinaisons paire et impaire, qu'on doit normaliser. On obtient donc:
Energie 2
*
2
1 2⎟⎠⎞
⎜⎝⎛=∞
LmE πh , 2
2
*
2
2n
LmEn ⎟
⎠⎞
⎜⎝⎛=∞ πh
Fonction d’onde dans le puits :
⎪⎪⎩
⎪⎪⎨
⎧
==
==
...6,4,2)sin(2)(
...5,3,1)cos(2)(
nzL
nL
r
nzL
nL
z
n
n
πϕ
πϕ
r
et nulle dans la barrière.
Puits quantique constitué d'une couche fine : On passe à trois dimensions, et on n'oublie pas la partie périodique de la fonction d'onde:
Energie 2
*
2
1 2⎟⎠⎞
⎜⎝⎛=∞
LmE πh , ( )22
*
22
2
*
2
0 22),,( yxyx kk
mn
LmEkknE ++⎟
⎠⎞
⎜⎝⎛+=
hh π
Fonction d’onde dans le puits : ( )
( )⎪⎪⎩
⎪⎪⎨
⎧
=+=
=+=
...6,4,2)(exp)sin()()(
...5,3,1)(exp)cos()()(
0
0
nykxkizL
nrur
nykxkizL
nrur
yxn
yxn
πψ
πψ
rr
rr
et nulle dans la barrière.
b. Effet de la hauteur finie des barrières
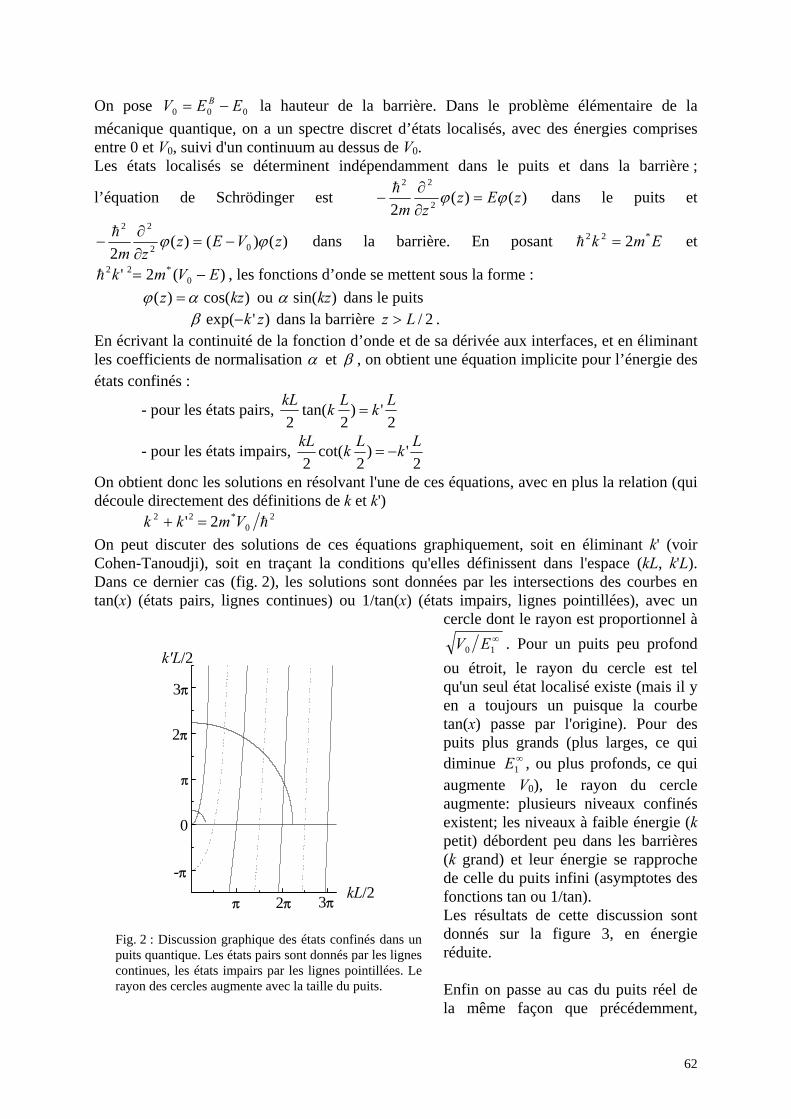
62
On pose 000 EEV B −= la hauteur de la barrière. Dans le problème élémentaire de la mécanique quantique, on a un spectre discret d’états localisés, avec des énergies comprises entre 0 et V0, suivi d'un continuum au dessus de V0. Les états localisés se déterminent indépendamment dans le puits et dans la barrière ;
l’équation de Schrödinger est )()(2 2
22
zEzzm
ϕϕ =∂∂
−h dans le puits et
)()()(2 02
22
zVEzzm
ϕϕ −=∂∂
−h dans la barrière. En posant Emk *22 2=h et
)(2' 0*22 EVmk −=h , les fonctions d’onde se mettent sous la forme :
)cos()( kzz αϕ = ou )sin(kzα dans le puits )'exp( zk−β dans la barrière 2/Lz > .
En écrivant la continuité de la fonction d’onde et de sa dérivée aux interfaces, et en éliminant les coefficients de normalisation α et β , on obtient une équation implicite pour l’énergie des états confinés :
- pour les états pairs, 2
')2
tan(2
LkLkkL=
- pour les états impairs, 2
')2
cot(2
LkLkkL−=
On obtient donc les solutions en résolvant l'une de ces équations, avec en plus la relation (qui découle directement des définitions de k et k')
20
*22 2' hVmkk =+ On peut discuter des solutions de ces équations graphiquement, soit en éliminant k' (voir Cohen-Tanoudji), soit en traçant la conditions qu'elles définissent dans l'espace (kL, k'L). Dans ce dernier cas (fig. 2), les solutions sont données par les intersections des courbes en tan(x) (états pairs, lignes continues) ou 1/tan(x) (états impairs, lignes pointillées), avec un
cercle dont le rayon est proportionnel à ∞10 EV . Pour un puits peu profond
ou étroit, le rayon du cercle est tel qu'un seul état localisé existe (mais il y en a toujours un puisque la courbe tan(x) passe par l'origine). Pour des puits plus grands (plus larges, ce qui diminue ∞
1E , ou plus profonds, ce qui augmente V0), le rayon du cercle augmente: plusieurs niveaux confinés existent; les niveaux à faible énergie (k petit) débordent peu dans les barrières (k grand) et leur énergie se rapproche de celle du puits infini (asymptotes des fonctions tan ou 1/tan). Les résultats de cette discussion sont donnés sur la figure 3, en énergie réduite. Enfin on passe au cas du puits réel de la même façon que précédemment,
k'L/2
kL/2
0
-π
3π
2π
2π
π
3π
π
Fig. 2 : Discussion graphique des états confinés dans un puits quantique. Les états pairs sont donnés par les lignes continues, les états impairs par les lignes pointillées. Le rayon des cercles augmente avec la taille du puits.
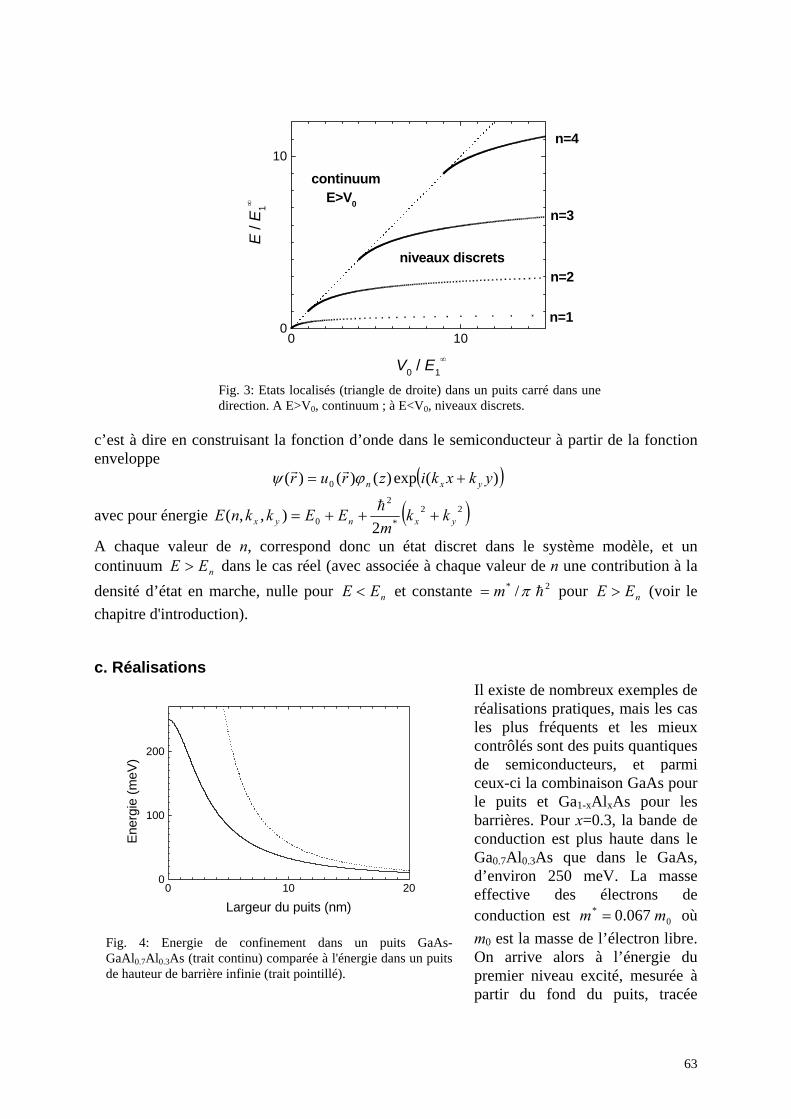
63
c’est à dire en construisant la fonction d’onde dans le semiconducteur à partir de la fonction enveloppe
( ))(exp)()()( 0 ykxkizrur yxn += ϕψ rr
avec pour énergie ( )22*
2
0 2),,( yxnyx kk
mEEkknE +++=
h
A chaque valeur de n, correspond donc un état discret dans le système modèle, et un continuum nEE > dans le cas réel (avec associée à chaque valeur de n une contribution à la densité d’état en marche, nulle pour nEE < et constante 2* / hπm= pour nEE > (voir le chapitre d'introduction).
c. Réalisations Il existe de nombreux exemples de réalisations pratiques, mais les cas les plus fréquents et les mieux contrôlés sont des puits quantiques de semiconducteurs, et parmi ceux-ci la combinaison GaAs pour le puits et Ga1-xAlxAs pour les barrières. Pour x=0.3, la bande de conduction est plus haute dans le Ga0.7Al0.3As que dans le GaAs, d’environ 250 meV. La masse effective des électrons de conduction est 0
* 067.0 mm = où m0 est la masse de l’électron libre. On arrive alors à l’énergie du premier niveau excité, mesurée à partir du fond du puits, tracée
0
10
0 10
n=3
n=2
n=1
n=4
niveaux discrets
continuum E>V0
V0 / E1∞
E /
E1∞
Fig. 3: Etats localisés (triangle de droite) dans un puits carré dans une direction. A E>V0, continuum ; à E<V0, niveaux discrets.
0 10 200
100
200
Ener
gie
(meV
)
Largeur du puits (nm)
Fig. 4: Energie de confinement dans un puits GaAs-GaAl0.7Al0.3As (trait continu) comparée à l'énergie dans un puits de hauteur de barrière infinie (trait pointillé).
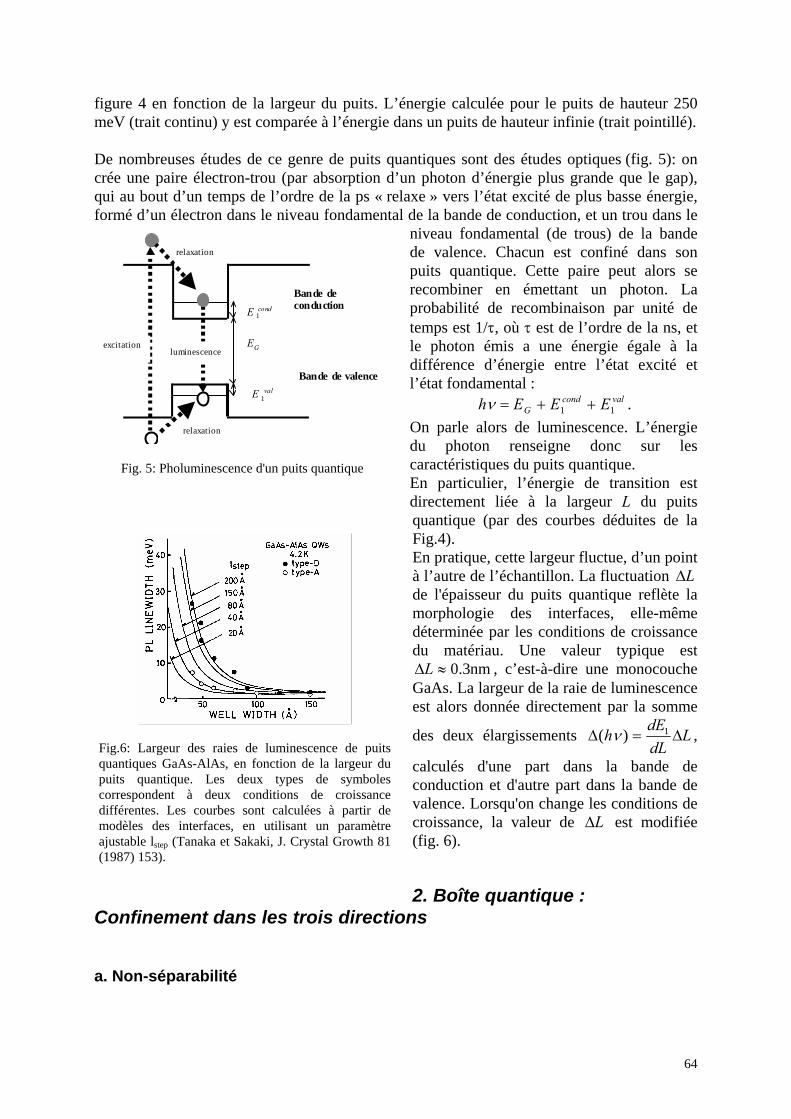
64
figure 4 en fonction de la largeur du puits. L’énergie calculée pour le puits de hauteur 250 meV (trait continu) y est comparée à l’énergie dans un puits de hauteur infinie (trait pointillé).
De nombreuses études de ce genre de puits quantiques sont des études optiques (fig. 5): on crée une paire électron-trou (par absorption d’un photon d’énergie plus grande que le gap), qui au bout d’un temps de l’ordre de la ps « relaxe » vers l’état excité de plus basse énergie, formé d’un électron dans le niveau fondamental de la bande de conduction, et un trou dans le
niveau fondamental (de trous) de la bande de valence. Chacun est confiné dans son puits quantique. Cette paire peut alors se recombiner en émettant un photon. La probabilité de recombinaison par unité de temps est 1/τ, où τ est de l’ordre de la ns, et le photon émis a une énergie égale à la différence d’énergie entre l’état excité et l’état fondamental :
valcondG EEEh 11 ++=ν .
On parle alors de luminescence. L’énergie du photon renseigne donc sur les caractéristiques du puits quantique. En particulier, l’énergie de transition est directement liée à la largeur L du puits quantique (par des courbes déduites de la Fig.4). En pratique, cette largeur fluctue, d’un point à l’autre de l’échantillon. La fluctuation L∆ de l'épaisseur du puits quantique reflète la morphologie des interfaces, elle-même déterminée par les conditions de croissance du matériau. Une valeur typique est
nm3.0≈∆L , c’est-à-dire une monocouche GaAs. La largeur de la raie de luminescence est alors donnée directement par la somme
des deux élargissements LdLdEh ∆=∆ 1)( ν ,
calculés d'une part dans la bande de conduction et d'autre part dans la bande de valence. Lorsqu'on change les conditions de croissance, la valeur de L∆ est modifiée (fig. 6).
2. Boîte quantique : Confinement dans les trois directions
a. Non-séparabilité
condE 1
GE
valE 1
Bande deconduction
Bande de valence
relaxation
relaxation
excitationluminescence
Fig. 5: Pholuminescence d'un puits quantique
Fig.6: Largeur des raies de luminescence de puits quantiques GaAs-AlAs, en fonction de la largeur du puits quantique. Les deux types de symboles correspondent à deux conditions de croissance différentes. Les courbes sont calculées à partir de modèles des interfaces, en utilisant un paramètre ajustable lstep (Tanaka et Sakaki, J. Crystal Growth 81 (1987) 153).

65
La plupart des réalisations expérimentales correspondent à une inclusion d’un matériau à petit gap, dans un matériau à plus grand gap. L’équation à résoudre est donc
( ) )()()()(2 0*
2
rFEErFrVrFm
rrrrh−=+∆−
où 0)( =rV r dans la boîte quantique (l’inclusion) et 0)( VrV =
r dans le matériau barrière. Dans le cas général, cette équation différentielle n’est pas à variables séparables, c’est-à-dire qu’on ne peut pas en chercher une solution sous forme d’un produit de trois fonctions d’une variable. En effet, même pour une boîte de forme cubique, le potentiel ne peut pas être écrit sous la forme d'une somme de trois potentiels, dont chacun correspondrait à une direction. On ne peut donc pas écrire l'hamiltonien comme une somme de trois opérateurs qui commutent.
On pourrait penser, néanmoins, qu'un produit de trois fonctions propres de puits quantiques de largeur L dans les trois directions x, y et z fournirait une solution du problème de la boîte cubique: eh non! car il s'agit de la fonction propre pour un potentiel 0)( =rV r pour x, y et z<L/2, 0)( VrV =
r pour x>L/2 et x,y<L/2 (et permutations), mais 02)( VrV =
r pour x,y>L/2 et z<L/2, etc..., ainsi que décrit fig. 7.
b. Cas de barrières infiniment hautes Le problème ne se pose évidemment plus si ∞→0V . L’équation différentielle se sépare en trois équations indépendantes, d’où des états caractérisés par trois entiers positifs, nml, d’énergie propre
⎥⎥
⎦
⎤
⎢⎢
⎣
⎡⎟⎟⎠
⎞⎜⎜⎝
⎛+⎟
⎟⎠
⎞⎜⎜⎝
⎛+⎟⎟
⎠
⎞⎜⎜⎝
⎛+=
222
*
2
0 2),,( l
Lm
Ln
LmElmnE
zyx
πππh ,
et de fonction enveloppe )()()()( zyxrF lmn ϕϕϕ=r .
On a donc des niveaux discrets. Le niveau fondamental est non-dégénéré (mais il faut ajouter la dégénérescence de spin) ; si les trois dimensions sont identiques (boîte cubique), le premier niveau excité a une dégénérescence triple ; et ainsi de suite... En variant les facteurs de forme, on pourra se poser le problème d'un fil carré, ou d'une galette carrée... On peut aller plus loin dans le problème d'un confinement avec des hauteurs de barrière finies, si on s'intéresse à des objets possédant des éléments de symétrie suffisants: on va s'intéresser au cas des symétries cylindrique et sphérique.
c. Confinement dans un disque. On suppose que le potentiel peut s'écrire ( ) ( ) ( )ρVzVrV +=
r , indépendant de θ en coordonnées cylindriques ( ) ( ) zyx ,sin,cos θρθρ == .
Fig. 7: A gauche, boîte quantique entre un matériau "boîte" de potentiel minimum (en blanc) et un matériau "barrière" de hauteur V0, en gris; à droite, le problème à variables séparables, avec en noir un matériau de hauteur 2V0.

66
Cette hypothèse est une bonne approche de plusieurs systèmes réels: - boîtes quantiques de semiconducteurs présentant un profil aplati (voir plus loin dans ce chapitre) - échantillon permettant l'étude du transport électronique à travers un pilier gravé dans un puits quantique (voir Pb DEA 2001) - "corral quantique" à la surface d'un métal,... L'hamiltonien de la fonction enveloppe s'écrit alors comme la somme de deux opérateurs qui commutent:
pz HHH += où )(2 *
2
zVm
pzz +=H et )(
2 *
22
ρVm
pp yxplan +
+=H .
On cherche donc les fonctions enveloppe sous la forme ( ) ( ) ( )ρrr gzfrF = . On peut utiliser l'expression du Laplacien en coordonnées cylindriques:
2
2
22
2
2
2 11θρρρρ ∂∂
+∂∂
+∂∂
+∂∂
=∆z
.
Il est évidemment tentant de définir, à ce point, deux opérateurs
( )ρρρρ
Vm
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+∂∂
−1
2 2
2
*
2h et 2
2
2*
2 12 θρ ∂
∂−
mh ; mais ces deux opérateurs ne commutent
pas à cause du terme radial du dernier. On va donc trouver un autre opérateur, qui commute avec zH et planH , et exprime la symétrie du problème lors d'une rotation d'angle θ quelconque. Un bon candidat est la dérivée partielle par rapport à θ. Et comme (il suffit de faire le changement de variable), xyyx ∂∂−∂∂=∂∂ θ , on prend plutôt
( ) zz Lpri =×=∂∂−rr
h θ , la composante suivant z du moment cinétique. Les trois observables qui commutent sont donc l'hamiltonien de confinement suivant z, zH , l'hamiltonien de confinement dans les directions du plan, planH , et la composante suivant l'axe du moment cinétique. Les états propres de Lz sont les solutions ( )θf de l'équation ffi lh =∂∂− θ , d'où ( ) ( )hlθθ if exp= , où hl m= avec m un entier relatif. On cherche alors les états propres de
planH sous la forme ( ) ( ) ( )θρρ imRg exp=r , qu'on introduit dans l'équation de Schrödinger de
planH pour obtenir l'équation dont R est solution:
REm
RRR plan2
*
2
2 2'"h
l=−+
ρρ.
Cette équation se réduit, si l'on pose hEmu *2ρ= et ( ) ( )uyR =ρ , à une équation sans dimension
01'" 2
2
=⎟⎟⎠
⎞⎜⎜⎝
⎛−++ y
um
uyy ,
qui gagne à être connue car elle se rencontre dans de nombreux problèmes à symétrie sphérique (oscillation d'un fil pesant, Bernoulli 1732; vibrations d'une membrane circulaire; modes électromagnétiques d'une cavité cylindrique; propagation dans un câble coaxial; perturbation des mouvements planétaires, Bessel 1824...). Il n'y a donc plus qu'à aller chercher les solutions dans un livre de fonctions mathématiques, qui nous dit qu'on doit prendre les combinaisons linéaires de deux fonctions dites de Bessel:

67
( )uJ m et ( )uYm , qui sont dûment tabulées. La fonction Jm est la seule solution bornée, alors que la fonction Ym diverge en u = 0. La fonction Jm est donnée fig. 8 pour les premières valeurs entières de m. On notera que J0 est la seule qui prenne une valeur non-nulle à l'origine. Dans le cas le plus général, on devra utiliser des combinaisons linéaires permettant la continuité de la fonction et de sa dérivée aux interfaces (comme dans le cas du puits quantique 2D de hauteur finie). Application: corral quantique. Le calcul ci-dessus permet une comparaison aux mesures de "corral quantique" tel que celui de la figure 9: des d'atomes de fer (Fe) ont été déposés sur la surface (111) d'un cristal de cuivre (Cu), et mis en place à l'aide d'une pointe STM (scanning tunneling microscope) pour former un cercle. La surface est ensuite étudiée à l'aide de la même pointe STM. Les atomes apparaissent comme un cercle de plots. Les ondes stationnaires à l'intérieur du "corral" sont attribuées aux états électroniques à la surface du cuivre. Ce sont ces états que l'on décrit dans la suite. On note z la normale à la surface, x et y deux directions du plan, 0ρ .le rayon du corral. L'hypothèse de travail est la suivante: (i) confinement dans la direction z: à la surface du cuivre, les électrons sont confinés dans l'état fondamental d'un puits de potentiel d'épaisseur L très faible (typiquement sur une distance inter-atomique à la surface du cuivre); les hauteurs de barrières sont très grandes; des mesures antérieures ont montré que le mouvement libre dans le plan x,y est alors décrit par une masse effective m* = 0.38 m0; (ii) confinement dans le plan x,y: les atomes de fer créent un potentiel de confinement latéral (directions x et y), en forme de disque de rayon R, également avec des barrières très hautes.
On a donc confinement dans un disque, et les deux potentiels (suivant z et dans le plan) sont très grands et sont donc considérés comme additifs. D'après ce qui précède, les fonctions enveloppe décrivant les électrons confinés dans le disque sont obtenues comme le produit d'une fonction de confinement en z, avec une énergie caractéristique eV 12 22 ≈≈ mLh (fig. 10 ; à l'échelle de la centaine de meV qui sera considérée, seul l'état de plus basse énergie est utile), et d'une fonction décrivant le mouvement
Fig. 8 : Fonctions de Bessel Jm pour les premières valeurs entières de m.
Fig. 9 : Corral quantique: confinement des électrons par un cercle d'atomes de Fe sur une surface de cuivre (Pour la Science, décembre 2001)
0 1 2 3m
Fig. 10 : Confinement dans le corral quantique; l'échelle de gauche est due au confinement en z et l'énergie caractéristique (double flèche) en est l'eV; l'échelle de droite correspond au mouvement dans le plan et l'échelle caractéristique est de quelques meV.

68
dans le plan, ( ) ( ) ( )θρρ imRg exp=r . Pour une valeur fixée de m, la fonction radiale s'écrit
( ) ( )uyR =ρ avec hEmu *2ρ= , et on a vu que c'est une combinaison des fonctions de Bessel. Dans le cas présent, elle ne peut pas contenir la fonction Ym puisque celle-ci diverge à l'origine. Donc ( ) ( )uJR m=ρ , à une constante multiplicative près. Comme les barrières sont) très hautes, la fonction R doit s'annuler sur le bord du corral ( 0ρρ = ), et hEmu *
00 2ρ= est donc un des zéros de la fonction de Bessel (valeurs de u telles que Jm(u) = 0). Les énergies propres pour le confinement du mouvement dans le plan sont donc données par
2
0
,*
2
, 2 ⎟⎟⎠
⎞⎜⎜⎝
⎛=
ρnm
nm
um
E h , où nmu , est le n-ième zéro de la fonction de Bessel Jm:
- pour J0 : u0,0 = 2.405, u0,1 = 5.520, u0,2 = 8.654, u0,3 = 11.79, u0,4 = 14.93, u0,5 = 18.07, etc... - pour J1 : u1,0 = 3.832, u1,1 = 7.016, u1,2 = 10.173, u1,3 = 13.32, u1,4 = 16.47 etc... Pour un corral de rayon ρ0 = 7.1 nm, en plaçant la pointe au centre du corral, M.F.Crommie,
C.P.Lutz, D.M.Eigler ont mesuré des accidents de la caractéristique électrique aux valeurs suivantes de V: -425 mV, -370 mV, -274 mV, -148 mV, 0, +170 mV (Science 262, 218 (1993)). Ils les ont attribués au passage en coïncidence du potentiel chimique de la pointe STM avec les niveaux électroniques confinés dans le corral.. Cette interprétation est testée sur la fig. 11, qui utilise les zéros de la fonction de Bessel J0 (la seule qui ne s'annule pas à l'origine et peut donc être contactée en plaçant la pointe STM au centre du corral). D'autres énergies caractéristiques apparaissent en effet lorsque la pointe est déplacée, en accord avec les zéros des autres fonctions de Bessel.
d. Système à symétrie sphérique Il s’agit d’un problème de « potentiel central »: parce que le système est à symétrie sphérique, on peut écrire l’équation différentielle en coordonnées sphériques (voir Cohen-Tannoudji) :
),,(),,()(),,(2
1),,(12
22*2
2
*
2
ϕθϕθϕθϕθ rEFrFrVrFLrm
rrFrrm
=++∂∂
−rh
où l’opérateur moment cinétique est défini par ∇×−=×=rr
hrrr
riprL Cette équation s’applique en tout point 0≠r . Le point r=0 doit être étudié à part. Les solutions se factorisent en une partie radiale )(rR et une partie angulaire qui est une harmonique sphérique ),( ϕθm
lY . L’harmonique sphérique est état propre de deux opérateurs:
le carré du moment cinétique 2Lr
, avec la valeur propre 2)1( h+ll , et la projection du moment cinétique zL , avec la valeur propre hm . La partie radiale est donc solution de
-500 -400 -300 -200 -100 0 100 200
0
100
200
300
400
500
600
700
Ener
gie
du n
ivea
u (m
eV)
Tension appliquée (mV)
Fig. 11 : Comparaison entre l'énergie calculée pour un électron confiné dans le corral de rayon 7.1 nm, et la tension de la pointe STM (mesures de M.F.Crommie, C.P.Lutz, D.M.Eigler, Science 262, 218 (1993))
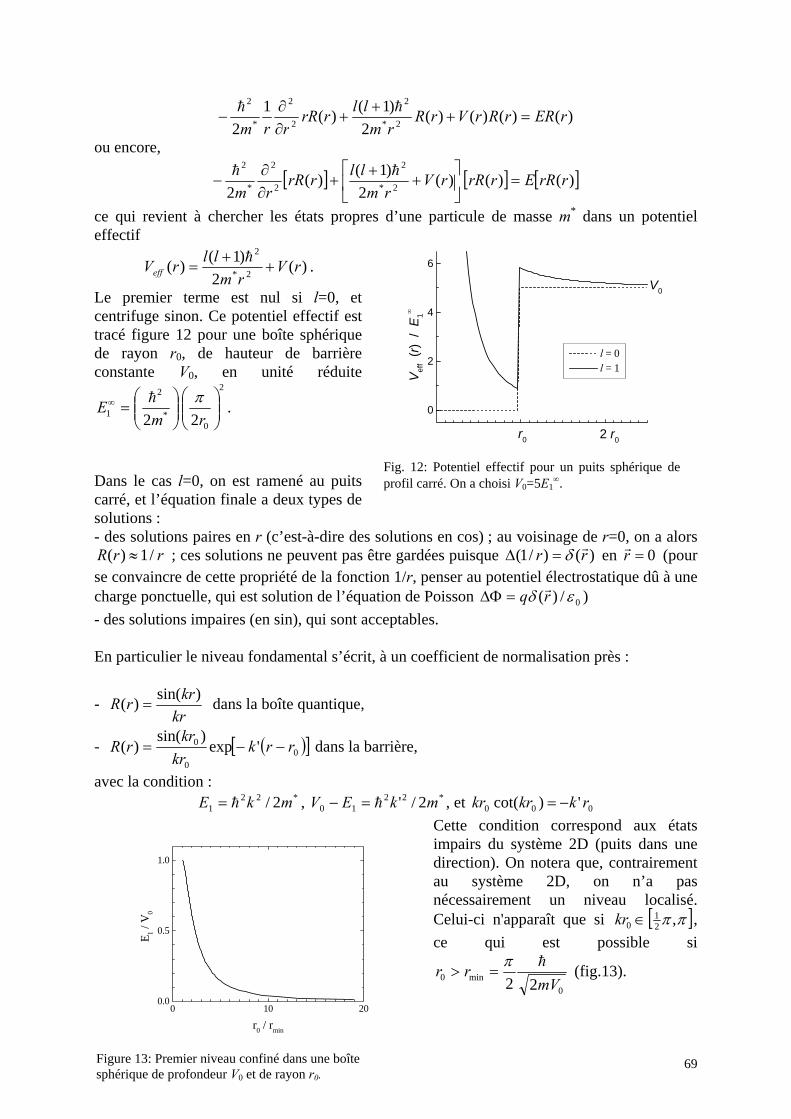
69
)()()()(2
)1()(12 2*
2
2
2
*
2
rERrRrVrRrm
llrrRrrm
=++
+∂∂
−hh
ou encore,
[ ] [ ] [ ])()()(2
)1()(2 2*
2
2
2
*
2
rrRErrRrVrm
llrrRrm
=⎥⎦
⎤⎢⎣
⎡+
++
∂∂
−hh
ce qui revient à chercher les états propres d’une particule de masse m* dans un potentiel effectif
)(2
)1()( 2*
2
rVrm
llrVeff ++
=h .
Le premier terme est nul si l=0, et centrifuge sinon. Ce potentiel effectif est tracé figure 12 pour une boîte sphérique de rayon r0, de hauteur de barrière constante V0, en unité réduite
2
0*
2
1 22 ⎟⎟⎠
⎞⎜⎜⎝
⎛⎟⎟⎠
⎞⎜⎜⎝
⎛=∞
rmE πh .
Dans le cas l=0, on est ramené au puits carré, et l’équation finale a deux types de solutions : - des solutions paires en r (c’est-à-dire des solutions en cos) ; au voisinage de r=0, on a alors
rrR /1)( ≈ ; ces solutions ne peuvent pas être gardées puisque )()/1( rr rδ=∆ en 0=rr (pour se convaincre de cette propriété de la fonction 1/r, penser au potentiel électrostatique dû à une charge ponctuelle, qui est solution de l’équation de Poisson 0/)( εδ rq r
=∆Φ ) - des solutions impaires (en sin), qui sont acceptables. En particulier le niveau fondamental s’écrit, à un coefficient de normalisation près :
- kr
krrR )sin()( = dans la boîte quantique,
- ( )[ ]00
0 'exp)sin(
)( rrkkr
krrR −−= dans la barrière,
avec la condition : *22
1 2/ mkE h= , *2210 2/' mkEV h=− , et 000 ')cot( rkkrkr −=
Cette condition correspond aux états impairs du système 2D (puits dans une direction). On notera que, contrairement au système 2D, on n’a pas nécessairement un niveau localisé. Celui-ci n'apparaît que si [ ]ππ ,2
10 ∈kr ,
ce qui est possible si
0min0 22 mV
rr hπ=> (fig.13).
0
2
4
6
V0
2 r0r0
l = 0 l = 1
Vef
f (r)
/ E
1∞
Fig. 12: Potentiel effectif pour un puits sphérique de profil carré. On a choisi V0=5E1
∞.
0.0
0.5
1.0
0 10 20
r0 / rmin
E 1 / V
0
Figure 13: Premier niveau confiné dans une boîte sphérique de profondeur V0 et de rayon r0.

70
Avec un peu de travail, on peut exprimer les états excités à l’aide des "fonctions de Bessel sphériques" (voir D.Bimberg, M.Grundmann et N.N.Ledentsov, Quantum dot heterostructures, Wiley 1999).
Dans le cas de barrières infinies, les énergies s'écrivent 2,
2
0
2
2 lnkrm ⎥⎦
⎤⎢⎣
⎡πh , où les lnk , sont les
zéros des fonctions de Bessel sphériques données dans le tableau suivant.
l=0 l=1 l=2 l=3 n=0 3.14 4.49 5.76 6.99 n=1 6.28 7.73 9.10 10.42 n=2 9.42
Les premiers niveaux sont schématisés sur la fig.14. Pour identifier un niveau, il suffit de spécifier la partie radiale et le module du moment cinétique. L'énergie du niveau ne dépend pas de la projection du moment cinétique. Il est traditionnel de noter ces niveaux en utilisant le nombre n de zéros de la fonction radiale (attention: pour les puits quantique, on aurait pu utiliser le nombre de zéros, mais la
tradition utilise plutôt le rang de la fonction, égal au nombre de zéros +1). Pour le moment cinétique, on utilise, comme en physique atomique, "s" pour l=0, "p" pour l=1, "d" pour l=2, etc... On a donc une succession de niveaux 0s, 0p, 0d, ..., 1s, 1p,... ns, np... (fig. 14). Attention: en physique atomique, on tire parti des dégénérescences accidentelles propres à l'atome d'hydrogène et la notation utilise comme nombre quantique principal l'énergie du niveau, d'où la succession 1s, (2s, 2p), (3s, 3p, 3d) etc... Dans une boîte quantique, les conditions d'existence et les dégénérescences n'ont bien sûr aucune raison d'être les mêmes que pour l'atome d'hydrogène, et on indique en général le nombre de zéros de la fonction radiale, et pas directement l'énergie comme en physique atomique.
e. Un cas simple: le potentiel harmonique 3D C'est un exemple intéressant car les calculs peuvent être poussés assez loin (en particulier lorsqu'on introduira plusieurs électrons dans la seconde partie de ce chapitre). On peut étudier le cas d'un potentiel harmonique dans les trois directions, soit comme combinaison de trois potentiels harmoniques unidimensionnels, soit comme système à potentiel central. Dans le premier cas, on écrit l'équation à résoudre sous la forme
),,(),,()(21),,(
22222*
2
2
2
2
2
2
*
2
zyxEFzyxFzyxmzyxFzyxm
=+++⎥⎦
⎤⎢⎣
⎡∂∂
+∂∂
+∂∂
− ωh
Chacun des trois oscillateurs harmoniques donne lieu une échelle d'états non dégénérés, d'énergie ( ) ωh2
1+n où n est le nombre de noeuds. La combinaison des trois oscillateurs conduit donc à des niveaux: (0,0,0), d'énergie ωh2
3 , (1,0,0), (0,1,0), et (0,0,1), d'énergie ωh2
5 ,... (h,k,l), d'énergie ,)2/3( ωh+++ lkh ...
Energie
Description par 3
Description par un oscillateur à symétrie sphérique
dégéné-
.
.
.
.
.
2s
1s
1p
1d
0d0p0s
1f
0f
Figure 14: Niveaux d'énergies d'une boîte sphérique infiniment profonde.
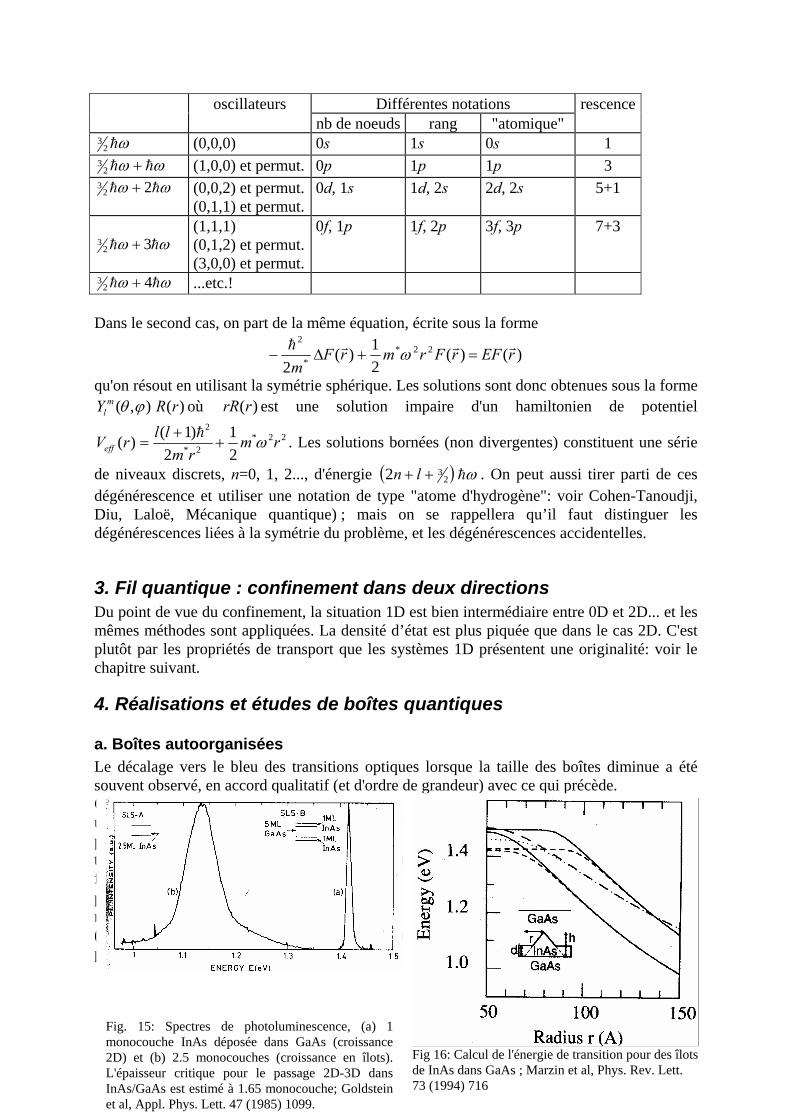
71
Différentes notations oscillateurs nb de noeuds rang "atomique"
rescence
ωh23 (0,0,0) 0s 1s 0s 1
ωω hh +23 (1,0,0) et permut. 0p 1p 1p 3
ωω hh 223 + (0,0,2) et permut.
(0,1,1) et permut. 0d, 1s 1d, 2s 2d, 2s 5+1
ωω hh 32
3 + (1,1,1) (0,1,2) et permut. (3,0,0) et permut.
0f, 1p 1f, 2p 3f, 3p 7+3
ωω hh 423 + ...etc.! Dans le second cas, on part de la même équation, écrite sous la forme
)()(21)(
222*
*
2
rEFrFrmrFm
rrrh=+∆− ω
qu'on résout en utilisant la symétrie sphérique. Les solutions sont donc obtenues sous la forme )(),( rRY m
l ϕθ où )(rrR est une solution impaire d'un hamiltonien de potentiel
22*2*
2
21
2)1()( rmrm
llrVeff ω++
=h . Les solutions bornées (non divergentes) constituent une série
de niveaux discrets, n=0, 1, 2..., d'énergie ( ) ωh232 ++ ln . On peut aussi tirer parti de ces
dégénérescence et utiliser une notation de type "atome d'hydrogène": voir Cohen-Tanoudji, Diu, Laloë, Mécanique quantique) ; mais on se rappellera qu’il faut distinguer les dégénérescences liées à la symétrie du problème, et les dégénérescences accidentelles.
3. Fil quantique : confinement dans deux directions Du point de vue du confinement, la situation 1D est bien intermédiaire entre 0D et 2D... et les mêmes méthodes sont appliquées. La densité d’état est plus piquée que dans le cas 2D. C'est plutôt par les propriétés de transport que les systèmes 1D présentent une originalité: voir le chapitre suivant.
4. Réalisations et études de boîtes quantiques
a. Boîtes autoorganisées Le décalage vers le bleu des transitions optiques lorsque la taille des boîtes diminue a été souvent observé, en accord qualitatif (et d'ordre de grandeur) avec ce qui précède. Cependant la forme des boîtes est en général complexe (par exemple, pyramides) : il faut utiliser des méthodes numériques pour calculer les niveaux confinés, et les dégénérescences prévues ci-dessus pour des formes très symétriques sont levées dans le cas réel. Il faut de plus tenir compte de l’effet des déformations élastiques. Mais le gros problème est celui des fluctuations de taille et de forme d’une boîte à l’autre. Même dans les cas des fluctuations les plus faibles (±10%), une mesure de luminescence sur un grand nombre de boîtes montre une raie très large (figures 15-17). Ce n'est qu'en limitant l’observation à un petit nombre de boîtes (élimination des autres par gravure chimique, ou méthode de microscopie optique en champ proche) que des raies très fines sont observées.
Fig. 15: Spectres de photoluminescence, (a) 1 monocouche InAs déposée dans GaAs (croissance 2D) et (b) 2.5 monocouches (croissance en îlots). L'épaisseur critique pour le passage 2D-3D dans InAs/GaAs est estimé à 1.65 monocouche; Goldstein et al, Appl. Phys. Lett. 47 (1985) 1099.
Fig 16: Calcul de l'énergie de transition pour des îlots de InAs dans GaAs ; Marzin et al, Phys. Rev. Lett. 73 (1994) 716
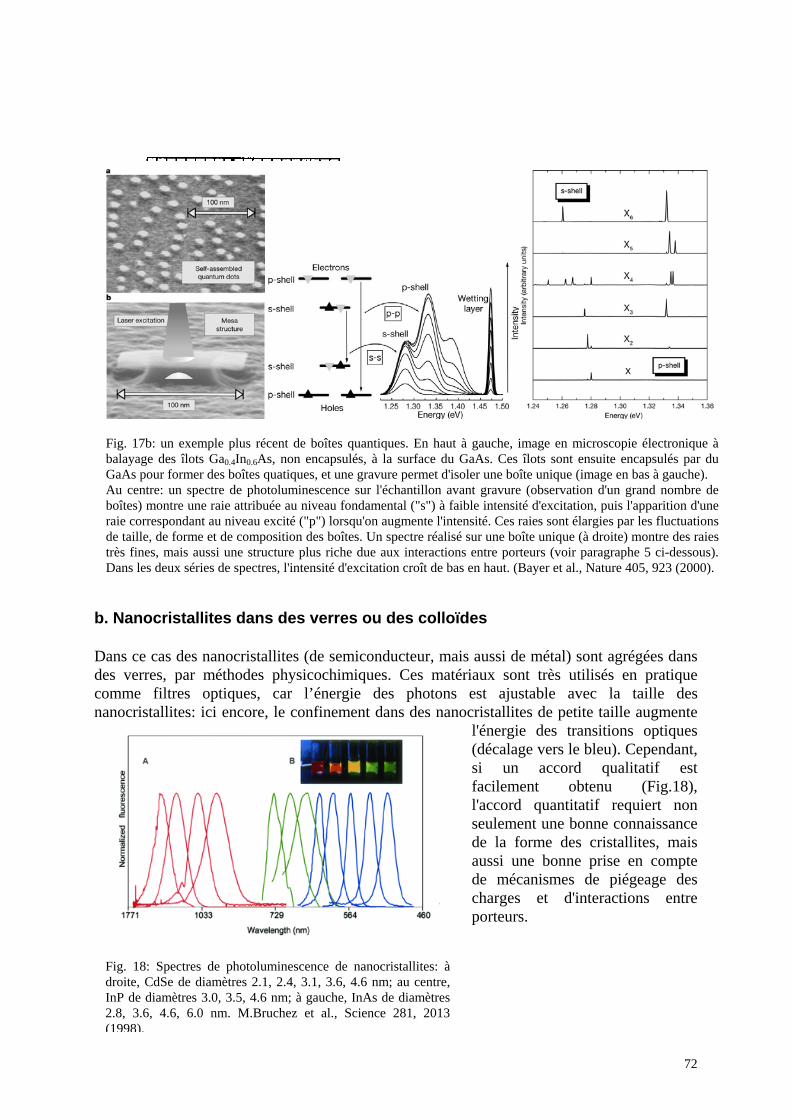
72
b. Nanocristallites dans des verres ou des colloïdes Dans ce cas des nanocristallites (de semiconducteur, mais aussi de métal) sont agrégées dans des verres, par méthodes physicochimiques. Ces matériaux sont très utilisés en pratique comme filtres optiques, car l’énergie des photons est ajustable avec la taille des nanocristallites: ici encore, le confinement dans des nanocristallites de petite taille augmente
l'énergie des transitions optiques (décalage vers le bleu). Cependant, si un accord qualitatif est facilement obtenu (Fig.18), l'accord quantitatif requiert non seulement une bonne connaissance de la forme des cristallites, mais aussi une bonne prise en compte de mécanismes de piégeage des charges et d'interactions entre porteurs.
Fig. 17a: Spectre de photoluminescence de boîtes quantiques InAs dans GaAs: une zone de dimension 500 nm (à gauche) ou 25 nm (à droite) a été délimitée sur l'échantillon pour diminuer le nombre de boîtes observées; (Marzin et al., Phys. Rev. Lett. 73,716 (1994))
Fig. 18: Spectres de photoluminescence de nanocristallites: à droite, CdSe de diamètres 2.1, 2.4, 3.1, 3.6, 4.6 nm; au centre, InP de diamètres 3.0, 3.5, 4.6 nm; à gauche, InAs de diamètres 2.8, 3.6, 4.6, 6.0 nm. M.Bruchez et al., Science 281, 2013 (1998).
Fig. 17b: un exemple plus récent de boîtes quantiques. En haut à gauche, image en microscopie électronique à balayage des îlots Ga0.4In0.6As, non encapsulés, à la surface du GaAs. Ces îlots sont ensuite encapsulés par du GaAs pour former des boîtes quatiques, et une gravure permet d'isoler une boîte unique (image en bas à gauche). Au centre: un spectre de photoluminescence sur l'échantillon avant gravure (observation d'un grand nombre de boîtes) montre une raie attribuée au niveau fondamental ("s") à faible intensité d'excitation, puis l'apparition d'une raie correspondant au niveau excité ("p") lorsqu'on augmente l'intensité. Ces raies sont élargies par les fluctuations de taille, de forme et de composition des boîtes. Un spectre réalisé sur une boîte unique (à droite) montre des raies très fines, mais aussi une structure plus riche due aux interactions entre porteurs (voir paragraphe 5 ci-dessous). Dans les deux séries de spectres, l'intensité d'excitation croît de bas en haut. (Bayer et al., Nature 405, 923 (2000).

73
5. Interactions entre électrons
a. Deux électrons dans une boîte quantique Jusqu’à maintenant, on s’est intéressé au cas où un seul électron est introduit dans le puits quantique ou la boîte quantique. Plus précisément, on a supposé que, parmi les quelques 106 électrons présents dans le volume de la boîte quantique (et les quelques 1021 dans le volume de l’échantillon), il y en a un dont le comportement peut-être décrit comme celui d’un électron unique, en présence du potentiel dû aux noyaux, aux autres électrons, aux champs extérieurs… Cette description en termes de modèle à un électron, parfois difficile à justifier (en particulier, l’ensemble des électrons présents devrait être décrit par une fonction d’onde antisymétrique, pour tenir compte du principe de Pauli qui s’applique à tout fermion…), est cependant très efficace : c'est dans de tels modèles à un électron (tight binding, pseudo-potentiels…) qu'on décrit les bandes de valence et de conduction d’un semi-conducteur, les bandes s et d d’un métal, etc…. On aboutit ainsi, dans le cas des semi-conducteurs, à la description utilisée ci-dessus d’un électron unique dans la bande de conduction en terme de masse effective et de fonction enveloppe. La même description s’adapte au cas d’un trou de la bande de valence. Des descriptions analogues sont utilisées dans les métaux, en terme de quasi-particules. Si plusieurs électrons de ce genre sont introduits (par exemple, dans la « bande de conduction » d’une boîte quantique de semi-conducteurs), l’interaction entre ces électrons doit être traitée complètement. On doit donc chercher les solutions antisymétriques d’une équation de Schrödinger (ou plutôt, de fonction enveloppe) à plusieurs électrons, comprenant le terme d’interaction entre ces électrons. Dans le cas de deux électrons, on a donc à résoudre
),,(),,(14
)()(22 22,1122,11
210
2
212*
2
1*
2
SrSrFESrSrFrr
qrVrVmm
rrrrrrrrrr
rrhh=
⎥⎥⎦
⎤
⎢⎢⎣
⎡
−+++∆−∆−
πεε
où les indices 1 et 2 désignent chacun des deux électrons. L'origine des énergies a été prise à 2E0, c'est à dire que dans l'équation ci-dessus, E remplace 02EE − . La fonction enveloppe F
dépend des coordonnées 1rr et 2r
r de chaque électron mais fait aussi intervenir leur spin 1Sr
ou
2Sr
. Elle doit être antisymétrique dans l’échange des deux électrons, puisque ce sont des fermions. Avant de rechercher une telle solution antisymétrique, on peut voir ce que donnent des approximations très simples et obtenir ainsi des ordres de grandeur. Tout d'abord, en l'absence d'interaction, la solution est obtenue en posant :
22112211 )()(),,,( SrfSrfSrSrF ba
rrrrrrrr=
où les fonctions ba ff , sont deux solutions de l'hamiltonien à un électron. En tenant compte de l'interaction à l'ordre le plus bas en perturbation, l'énergie des deux électrons est 0
abba EEE ∆++ où
23
132
12
1210
20 )()(1
4rdrdrfrf
rrqE baab
rrrrrr∫∫ −
=∆πεε
.

74
Fig. 19: Potentiel carré pour le premier électron (à gauche), et potentiel effectif pour le deuxième électron en tenant compte de la présence du premier (à droite); la figure du bas est un agrandissement (Palun, Lamouche et Fishman, Solid State Electronics 43, 1147 (1999))
Dans une boîte de largeur L dans les trois directions, l'ordre de grandeur sera Lq 02 4πεε , soit
meV14≈ si nm10=L et 10=ε . Une démarche un peu plus élaborée permet d’avoir une idée de la modification des fonctions d’onde ; elle consiste à rechercher l'état d'un électron en tenant compte de la présence de l'autre électron, celui-ci étant dans l'état non perturbé. Le premier électron est donc dans l'état a de fonction enveloppe af ,
solution de )()()(2 *
2
rfErfrVm
rrrh=⎥
⎦
⎤⎢⎣
⎡+∆− .
La probabilité de présence de cet électron permet de calculer la densité de charge qui lui est associée, 2)()( rfqr a
rr−=ρ . Cette densité
de charge donne lieu à un potentiel électrostatique )(rrΦ que l'on peut calculer (fig. 19) en intégrant l'équation de Poisson
0/)()( εερ rr rr=∆Φ . Enfin l'état du second
électron est calculé comme solution )(~ rfbr de la
nouvelle "équation de Schrödinger" )(~)(~)()(2 *
2
rfErfrqrVm
rrrrh=⎥
⎦
⎤⎢⎣
⎡Φ−+∆− .
On tient compte alors du principe d'exclusion de Pauli en imposant à un état avec a=b d'avoir les deux spins opposés. Ces deux calculs permettent d’avoir facilement des idées sur l’ordre de grandeur et le type de modification des fonctions d’onde, mais aucun n’est très satisfaisant. Dans le cas de deux électrons, on peut parfois calculer sans trop de difficulté les états symétriques et antisymétriques obéissant réellement au principe de Pauli. En premier, il est facile de tenir compte des spins. Deux spins ½ se combinent en :
- un état singulet de spin total 0=S , de fonction d’onde ( )21212
1 +−−−+ qui est antisymétrique dans l’échange des deux électrons. - un niveau triplet de spin total 1=S ; les fonctions d’onde s’écrivent :
( )21
212121
21
−−
+−+−+
++
et elles sont symétriques (invariantes) dans l’échange des deux électrons. La fonction totale, antisymétrique, s’obtient donc en multipliant la fonction de spin par une partie orbitale qui doit être symétrique dans le cas des états singulets de spin, et antisymétrique dans le cas des états triplets. Dans un tel système à deux particules, il est souvent utile de passer dans le repère du centre de masse. On pose de façon générale, pour deux particules de masses m1 et m2:
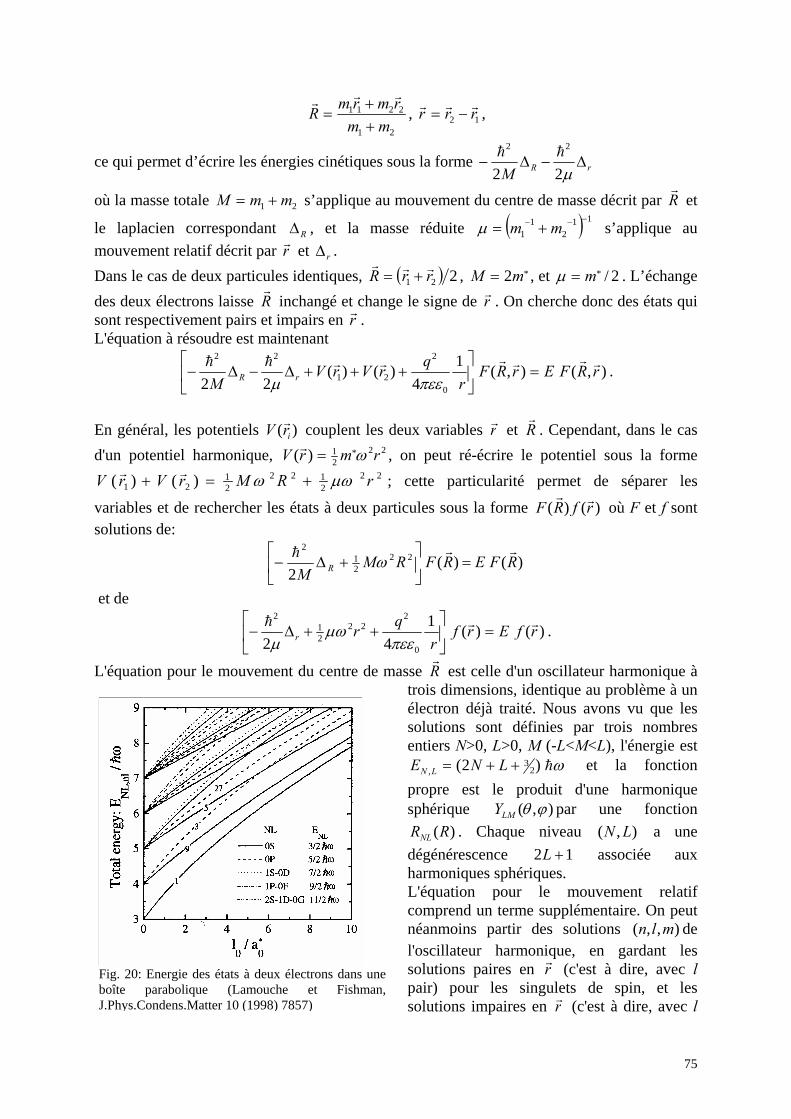
75
21
2211
mmrmrmR
++
=rrr
, 12 rrr rrr−= ,
ce qui permet d’écrire les énergies cinétiques sous la forme rRM∆−∆−
µ22
22 hh
où la masse totale 21 mmM += s’applique au mouvement du centre de masse décrit par Rr
et
le laplacien correspondant R∆ , et la masse réduite ( ) 112
11
−−− += mmµ s’applique au mouvement relatif décrit par rr et r∆ . Dans le cas de deux particules identiques, ( ) 221 rrR rrr
+= , *2mM = , et 2/*m=µ . L’échange des deux électrons laisse R
r inchangé et change le signe de rr . On cherche donc des états qui
sont respectivement pairs et impairs en rr . L'équation à résoudre est maintenant
),(),(14
)()(22 0
2
21
22
rRFErRFr
qrVrVM rR
rrrrrrhh=⎥
⎦
⎤⎢⎣
⎡+++∆−∆−
πεεµ.
En général, les potentiels )( irV r couplent les deux variables rr et R
r. Cependant, dans le cas
d'un potentiel harmonique, 22*21)( rmrV ω=
r , on peut ré-écrire le potentiel sous la forme 22
2122
21
21 )()( rRMrVrV µωω +=+rr
; cette particularité permet de séparer les
variables et de rechercher les états à deux particules sous la forme )()( rfRF rr où F et f sont
solutions de:
)()(2
2221
2
RFERFRMM R
rrh=⎥
⎦
⎤⎢⎣
⎡+∆− ω
et de
)()(142 0
222
21
2
rfErfr
qrrrrh
=⎥⎦
⎤⎢⎣
⎡++∆−
πεεµω
µ.
L'équation pour le mouvement du centre de masse Rr
est celle d'un oscillateur harmonique à trois dimensions, identique au problème à un électron déjà traité. Nous avons vu que les solutions sont définies par trois nombres entiers N>0, L>0, M (-L<M<L), l'énergie est
ωh)2( 23
, ++= LNE LN et la fonction propre est le produit d'une harmonique sphérique ),( ϕθLMY par une fonction
)(RRNL . Chaque niveau ),( LN a une dégénérescence 12 +L associée aux harmoniques sphériques. L'équation pour le mouvement relatif comprend un terme supplémentaire. On peut néanmoins partir des solutions ),,( mln de l'oscillateur harmonique, en gardant les solutions paires en rr (c'est à dire, avec l pair) pour les singulets de spin, et les solutions impaires en rr (c'est à dire, avec l
Fig. 20: Energie des états à deux électrons dans une boîte parabolique (Lamouche et Fishman, J.Phys.Condens.Matter 10 (1998) 7857)

76
impair) pour les triplets de spin. Le terme d'interaction Coulombienne se traite alors numériquement: il est suffisant d'évaluer un petit nombre de termes (Lamouche et Fishman, J.Phys.Condens.Matter 10 (1998) 7857). De plus, comme la symétrie sphérique est conservée pour l'équation dont )(rf r est solution, la dégénérescence associée aux harmoniques sphérique n'est pas levée. Les résultats du calcul sont donnés sur la figure 20 pour les premiers niveaux ( )lnLN ,;, . Les notations utilisent le nombre de noeuds de la fonction radiale. Le paramètre utilisé comme abscisse est le rapport de deux longueurs caractéristiques: [ ] 2/1
0 ωml h= pour l'oscillateur harmonique, et 22
0*0 4 mqa hπεε= pour l'interaction Coulombienne.
Le niveau fondamental est ( )0,0;0,0 , à l'énergie 0,03 E∆+ωh , où le décalage 0,0E∆ est dû à l'interaction entre les deux électrons dont le mouvement relatif est décrit par l'état
)0,0(),( =ln ; puisque l est pair, c'est un singulet de spin; la dégénérescence totale est 1. Le premier niveau excité est ( )1,0;0,0 , à l'énergie 1,04 E∆+ωh ; c'est un triplet de spin, et la dégénérescence totale est donc 9; le niveau ( )0,0;1,0 est à l'énergie 0,04 E∆+ωh , c'est un singulet de spin et la dégénérescence totale est 3. Les niveaux suivants se traitent de la même façon, bien que la complexité augmente rapidement... Il n'est pas inintéressant de reprendre ces trois niveaux dans le langage des électrons indépendants (fig. 21). On note alors ( )ln, les niveaux à un électron. En l'absence d'interaction, on a le niveau où les deux électrons sont dans ( )0,0 à l'énergie ωh3 , et à l'énergie ωh4 le niveau où un électron est dans ( )0,0 et un électron dans ( )1,0 . Dans le premier cas, les spins des deux électrons sont antiparallèles et la dégénérescence totale est 1. Dans le second cas, les états électroniques sont différents, tous les états de spin sont donc possibles et la dégénérescence totale est donc 223 ×× . L'état où les spins des deux électrons sont parallèles (c'est à dire l'état triplet) a une énergie plus basse: cela revient à noter que 0,0E∆ est plus grand que 1,0E∆ , ce qui se justifie bien par le fait que l'état de mouvement relatif ( )0,0 pour deux électrons sans interaction est le seul ayant une probabilité de présence non nulle en
0=r (les deux électrons au même endroit), ce qui va renforcer leur interaction dans un
ωh4
ωh3
00E∆
01E∆00E∆
00,0;1,0 =S
11,0;0,0 =S
00,0;0,0 =S
Fig 21: schéma des premiers niveaux pour deux électrons confinés.
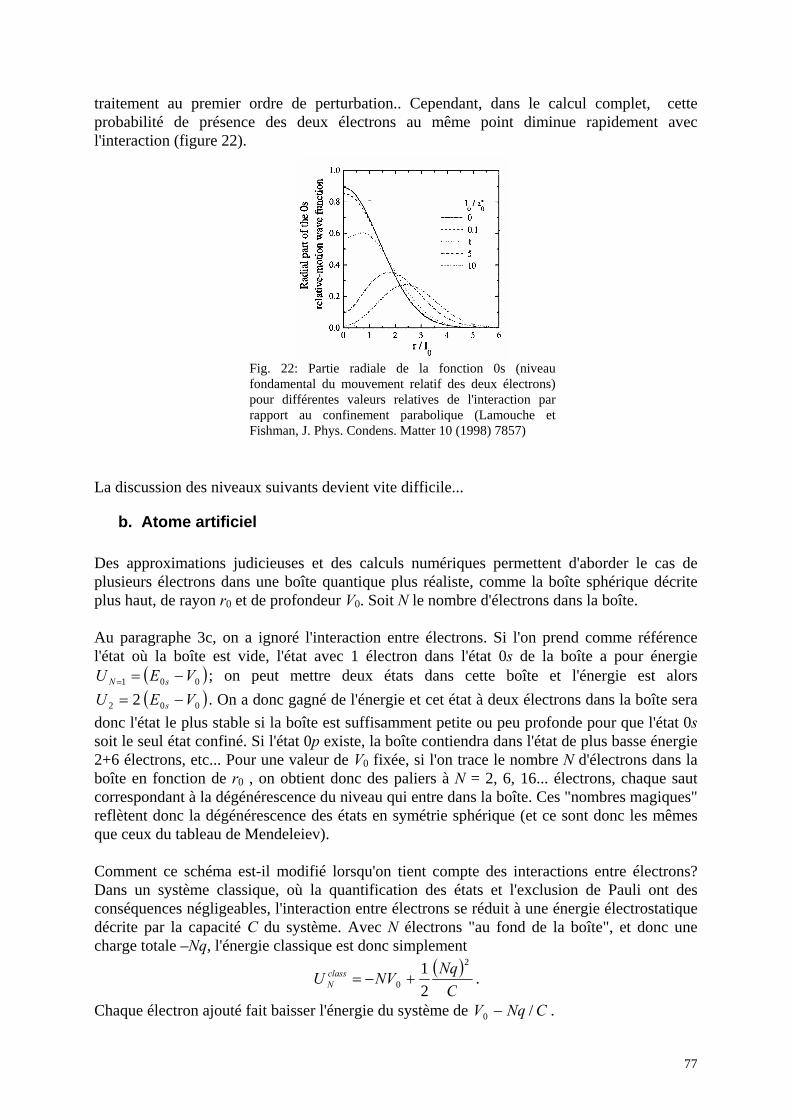
77
traitement au premier ordre de perturbation.. Cependant, dans le calcul complet, cette probabilité de présence des deux électrons au même point diminue rapidement avec l'interaction (figure 22).
La discussion des niveaux suivants devient vite difficile...
b. Atome artificiel Des approximations judicieuses et des calculs numériques permettent d'aborder le cas de plusieurs électrons dans une boîte quantique plus réaliste, comme la boîte sphérique décrite plus haut, de rayon r0 et de profondeur V0. Soit N le nombre d'électrons dans la boîte. Au paragraphe 3c, on a ignoré l'interaction entre électrons. Si l'on prend comme référence l'état où la boîte est vide, l'état avec 1 électron dans l'état 0s de la boîte a pour énergie
( )001 VEU sN −== ; on peut mettre deux états dans cette boîte et l'énergie est alors ( )002 2 VEU s −= . On a donc gagné de l'énergie et cet état à deux électrons dans la boîte sera
donc l'état le plus stable si la boîte est suffisamment petite ou peu profonde pour que l'état 0s soit le seul état confiné. Si l'état 0p existe, la boîte contiendra dans l'état de plus basse énergie 2+6 électrons, etc... Pour une valeur de V0 fixée, si l'on trace le nombre N d'électrons dans la boîte en fonction de r0 , on obtient donc des paliers à N = 2, 6, 16... électrons, chaque saut correspondant à la dégénérescence du niveau qui entre dans la boîte. Ces "nombres magiques" reflètent donc la dégénérescence des états en symétrie sphérique (et ce sont donc les mêmes que ceux du tableau de Mendeleiev). Comment ce schéma est-il modifié lorsqu'on tient compte des interactions entre électrons? Dans un système classique, où la quantification des états et l'exclusion de Pauli ont des conséquences négligeables, l'interaction entre électrons se réduit à une énergie électrostatique décrite par la capacité C du système. Avec N électrons "au fond de la boîte", et donc une charge totale –Nq, l'énergie classique est donc simplement
( )C
NqNVU classN
2
0 21
+−= .
Chaque électron ajouté fait baisser l'énergie du système de CNqV /0 − .
Fig. 22: Partie radiale de la fonction 0s (niveau fondamental du mouvement relatif des deux électrons) pour différentes valeurs relatives de l'interaction par rapport au confinement parabolique (Lamouche et Fishman, J. Phys. Condens. Matter 10 (1998) 7857)
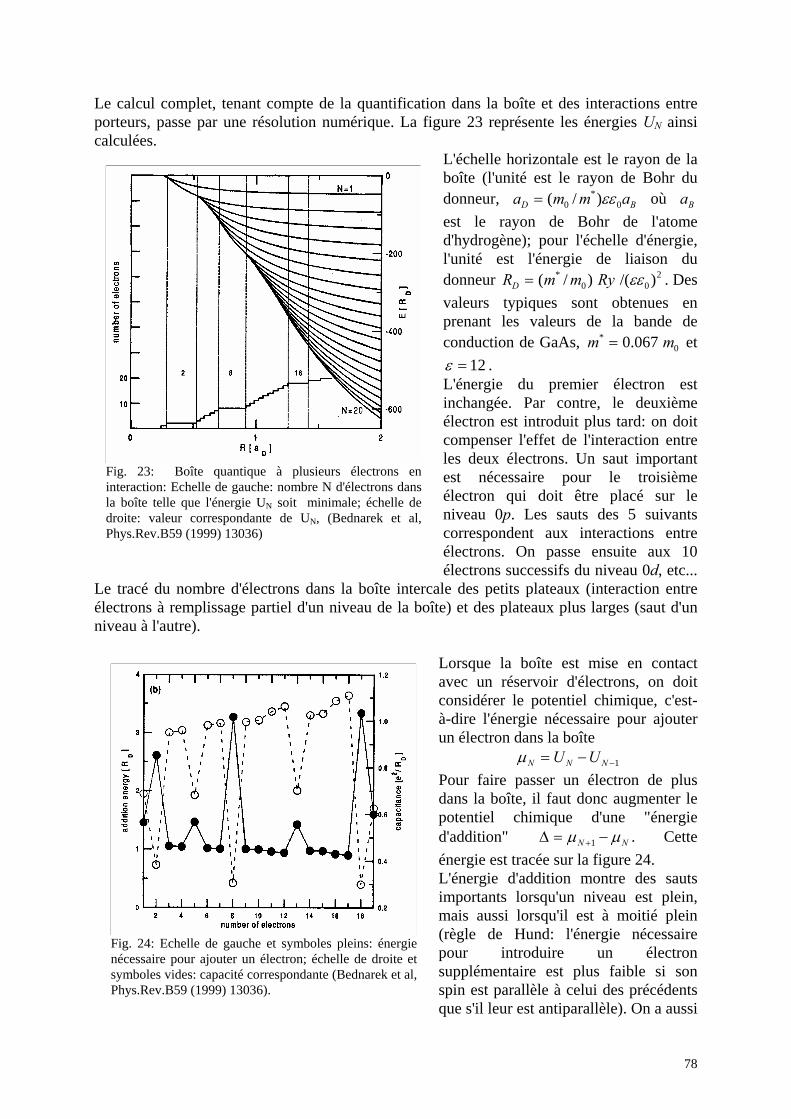
78
Le calcul complet, tenant compte de la quantification dans la boîte et des interactions entre porteurs, passe par une résolution numérique. La figure 23 représente les énergies UN ainsi calculées.
L'échelle horizontale est le rayon de la boîte (l'unité est le rayon de Bohr du donneur, BD amma 0
*0 )/( εε= où Ba
est le rayon de Bohr de l'atome d'hydrogène); pour l'échelle d'énergie, l'unité est l'énergie de liaison du donneur 2
00* )/()/( εεRymmRD = . Des
valeurs typiques sont obtenues en prenant les valeurs de la bande de conduction de GaAs, 0
* 067.0 mm = et 12=ε .
L'énergie du premier électron est inchangée. Par contre, le deuxième électron est introduit plus tard: on doit compenser l'effet de l'interaction entre les deux électrons. Un saut important est nécessaire pour le troisième électron qui doit être placé sur le niveau 0p. Les sauts des 5 suivants correspondent aux interactions entre électrons. On passe ensuite aux 10 électrons successifs du niveau 0d, etc...
Le tracé du nombre d'électrons dans la boîte intercale des petits plateaux (interaction entre électrons à remplissage partiel d'un niveau de la boîte) et des plateaux plus larges (saut d'un niveau à l'autre).
Lorsque la boîte est mise en contact avec un réservoir d'électrons, on doit considérer le potentiel chimique, c'est-à-dire l'énergie nécessaire pour ajouter un électron dans la boîte
1−−= NNN UUµ Pour faire passer un électron de plus dans la boîte, il faut donc augmenter le potentiel chimique d'une "énergie d'addition" NN µµ −=∆ +1 . Cette énergie est tracée sur la figure 24. L'énergie d'addition montre des sauts importants lorsqu'un niveau est plein, mais aussi lorsqu'il est à moitié plein (règle de Hund: l'énergie nécessaire pour introduire un électron supplémentaire est plus faible si son spin est parallèle à celui des précédents que s'il leur est antiparallèle). On a aussi
Fig. 23: Boîte quantique à plusieurs électrons en interaction: Echelle de gauche: nombre N d'électrons dans la boîte telle que l'énergie UN soit minimale; échelle de droite: valeur correspondante de UN, (Bednarek et al, Phys.Rev.B59 (1999) 13036)
Fig. 24: Echelle de gauche et symboles pleins: énergie nécessaire pour ajouter un électron; échelle de droite et symboles vides: capacité correspondante (Bednarek et al, Phys.Rev.B59 (1999) 13036).

79
tracé ∆= /2qC . En effet, pour un système classique,
CNqNVU class
N
2
0)(
21
+−=
ce qui permet de calculer
CNq
CqVcl
N
22
0 2+−−=µ et Cqclass /2=∆ .
On voit que la notion de capacité, qui décrit l'interaction entre porteurs dans le système classique, doit être précisée dans le cas de "boîtes quantiques". Les modifications mettent en jeu la quantification de l'énergie sur des niveaux, la dégénérescence de ces niveaux, et même la règle de Hund. Cette similitude avec les résultats de la physique atomique justifie le terme "atome artificiel" souvent utilisé dans ce cadre. On comprend aussi la différence entre le comportement des boîtes réalisées à base de métaux et celles réalisées avec des semiconducteurs. Dans le cas des semiconducteurs, il y a peu d'électrons dans la bande de conduction, et donc quelques électrons dans une boîte, sur des états faiblement dégénérés et dans des états quantiques à faible nombre de noeuds. Donc on saute souvent d'un niveau à l'autre et les énergies et fonctions d'onde sont fortement modifiés par l'interaction entre porteurs. Dans les métaux, le niveau de Fermi est dans la bande (on a de l'ordre d'un "électron libre" par atome du métal), donc quelques 103-10'4 électrons par boîte, sur des niveaux fortement dégénérés sur des états avec de nombreux noeuds. L'approche "électrostatique classique" restera utilisable sauf pour des objets extrêmement petits (agrégats métalliques).
Exercices
I. Puits quantique (système 2D): quelques évaluations en calcul de perturbation On considère un puits quantique GaAs-Ga1-xAlxAs d'épaisseur nm 10=L , et on s'intéresse à la bande de conduction: décalage de bande de conduction meV 2500 =V , masse effective
0* 067.0 mm = où kg 109 31
0−×=m est la masse de l'électron dans le vide. On néglige la
variation de masse dans la barrière 1. Calculer les énergies de confinement dans l'hypothèse d'une barrière de hauteur infinie. L'unité usuelle est le meV. 2.Ré-établir l'équation donnant l'énergie de confinement dans le niveau fondamental pour des barrières de hauteur finie, dans le problème à une dimension z (direction normale au puits); réécrire explicitement la fonction propre. 3. Dans le problème réel, réécrire explicitement les fonctions enveloppe en rr , et les fonctions d'onde complète, dans la première sous-bande (associée au (2)). Calculer la densité d'états (unité: cm-2 meV-1). 4. Esquissez une résolution graphique de l'équation du (2); donner les allures dans les cas limites (puits large et profond, puits étroit et peu profond) 5. Calculer la variation d'énergie E∆ pour une fluctuation L∆ de l'épaisseur du puits. Comparer à la probabilité de présence à l'interface ... et commenter. Application: L∆ = 1 monocouche GaAs pour un puits élaboré sur un substrat d'orientation (001). On donne le paramètre de maille cubique de GaAs, nm 565.00 =a . 6. Calculer la variation d'énergie E∆ pour une fluctuation 0V∆ de la hauteur de barrière. Comparer à la probabilité de présence intégrée sur les barrières ... et commenter. Application,

80
meV 100 =∆V , ce qui est l'ordre de grandeur qui correspond à une variation de l'ordre de 1% de la composition d'un alliage Ga1-xAlxAs
II. Boîte aplatie On considère une boîte quantique d'épaisseur nm 2=zL et de base carrée de largeur
nm 20== yx LL . On admettra dans un premier temps (et on justifiera ensuite) que dans un tel cas où yxz LLL ,<< , une approximation raisonnable consiste à simplement ajouter les énergies de confinement calculées pour les puits de hauteur finie dans chacune des trois directions. On effectue sur cet échantillon deux types de mesures: - des mesures de photoluminescence dans le domaine visible – proche infrarouge, où la transition consiste à faire passer un électron du niveau fondamental dans la bande de conduction au niveau fondamental des trous dans la bande de valence - des mesures en fréquence micro-ondes où on fait passer un électron du niveau fondamental dans la bande de conduction au premier niveau excité. Dans chaque mesure, on enregistre le signal correspondant à un grand nombre de boîtes. Discuter (qualitativement, en s'aidant des figures du cours) l'influence, sur les largeurs de raies de chaque expérience, des fluctuations de une monocouche de: - la largeur des boîtes - l'épaisseurs des boîtes ou pour des variations de 10 meV de la hauteur de barrière dues à une fluctuation de: - la composition du matériau des boîtes (fluctuations du gap) - la composition du matériau barrière (idem).
III. Test expérimental de la fonction enveloppe d'un puits quantique. 1. On considère un puits quantique formé d'une couche fine (16 nm) du semi-conducteur GaAs insérée dans des barrières Ga1-xAlxAs, et on s'intéresse aux électrons de la bande de conduction. Le décalage de la bande de conduction entre les deux matériaux pour la composition choisie est de 250 meV. On fait l'approximation d'une masse effective identique dans les deux matériaux, m*=0.067 m0. Rappeler rapidement l'application de la méthode de la fonction enveloppe à ce système. 2. On insère une couche fine (0.24 nm) de matériau Ga1-yInyAs à une position z par rapport au milieu du puits quantique. Ce matériau est un semi-conducteur à gap direct, dont le bas de la bande de conduction est situé 100 meV au dessous de celui de GaAs (pour la composition choisie). Là encore, on fait l'approximation d'une masse effective unique. Ecrire l'équation différentielle permettant de calculer les niveaux de confinement des électrons. Schématiser le potentiel adapté au problème. 3. Utiliser une méthode de perturbation pour calculer les déplacement des niveaux d'énergie dus à l'insertion de la couche Ga1-xInxAs. 4. Commenter la figure ci-dessous, issue de l'article "Experimental probing of quantum well eigenstates", J.-Y. Marzin et J.-M. Gérard, Phys. Rev. Lett. 62, 2172 (1989).

81
5. Quelle est l'influence d'une erreur systématique sur la quantité d'indium insérée?
Experimental probability density envelope ρ for (a) E1, (b) E2, (c) E3, compared to the theoretical effective mass result. Dots and triangles indicate experimental results obtained on two different samples.

82
Annexe:
La méthode de la fonction enveloppe: description et justification rapides L'approximation à un électron de la description des électrons dans un solide cristallin parfait
et infini conduit à rechercher les états propres de l'Hamiltonien crist0
2
2V
m+∆−
h où m0 est la
masse de l'électron et Vcrist est le potentiel "cristallin", périodique, dû aux ions du réseau et à tous les autres électrons. Les solutions sont fonctions de Bloch, qui s'écrivent sous la forme
( ) ( ) ( )rkirur kk
rrrrrr .exp=ψ où ( )ruk
rr est une fonction périodique et où le vecteur d'onde k
r prend
ses valeurs dans la première zone de Brillouin. Au voisinage d'un minimum de bande, l'énergie peut être décrite dans une approximation au second ordre en k par l'expression *22
0 2mkEEk hr += , qui définit la masse effective m* comme une simple paramétrisation de la courbure de la bande au voisinage de son minimum. On a supposé ici, pour simplifier, que le minimum est en k=0 et que la courbure est isotrope: c'est le cas pour des semi-conducteurs à gap direct tels que GaAs (pour lequel m*=0.067m0); pour d'autres matériaux, il faudra compliquer la description... On a également omis un second indice qui devrait spécifier la bande à laquelle on s'intéresse. Cette démarche permet de représenter les états propres du cristal parfait, infini, en l'absence de toute interaction avec un champ extérieur... On a en général à décrire le matériau en présence d'une perturbation, qui peut être un potentiel extérieur (électrostatique...), un défaut, une impureté, une interface, etc... Supposons que l'Hamiltonien décrivant un électron est
maintenant ( )rVVm
rh++∆− crist
0
2
2. On va s'intéresser aux états proches du minimum de la
bande. On cherche donc les nouvelles fonctions propres sous la forme ( ) ( ) ( )rFrur rrr0=ϕ , où
( )ru r0 est la partie périodique de la fonction de Bloch pour k=0; la fonction ( )rF r ainsi définie
est appelée "fonction enveloppe". En l'absence du potentiel de perturbation ( )rV r , la fonction enveloppe est F=1 pour la fonction de Bloch en k=0, et elle est proche d'une onde plane de grande longueur d'onde si k est proche de 0. On va donc faire l'hypothèse que l'effet de ( )rV r est "faible", si bien que la fonction enveloppe est peu localisée dans l'espace réel. De façon équivalente, la transformée de Fourier
( ) ( ) rdrkirFV
Fk3.exp1 rrr
r −= ∫ est "piquée" au voisinage de k=0, et kFr est nul si k est grand (à
l'échelle de la première zone de Brillouin, c'est-à-dire à l'échelle de 1/a où a est la maille élémentaire). Cette hypothèse est essentielle, elle devra être vérifiée en fin de calcul.
Lorsqu'on cherche à résoudre l'équation de Schrödinger, ϕϕ EVVmP
=⎟⎟⎠
⎞⎜⎜⎝
⎛++ crist*
2
2, on
peut utiliser le fait que l'action des deux premiers termes (l'hamiltonien non perturbé) sur les fonctions de Bloch est connue, et l'expliciter. Pour cela, on peut écrire
( ) ( ) ( ) ( ) ( )rueFrueFrurFr krki
kk
rki
kk
rrrrrr
rr
rr
rr
rr
.0
.0 ∑∑ ≈==ϕ
Cette dernière approximation parce que kuu r≈0 si k est petit et 0≈kFr sinon. On a donc
obtenu un développement de la fonction recherchée, ( ) ( )rFr kk
k
rrr
rr ψϕ ∑= , où ( )rk
rrψ est une

83
fonction de Bloch, état propre de l'hamiltonien non perturbé, de valeur propre *22
0 2mkEEk hr += . On l'écrit:
( ) ( )
( )
( )rEmkF
rVm
F
rFVm
rVm
kk
k
kk
k
kk
k
rh
rh
rhrh
rr
r
rr
r
rr
r
ψ
ψ
ψϕ
⎟⎟⎠
⎞⎜⎜⎝
⎛+=
⎟⎟⎠
⎞⎜⎜⎝
⎛+∆−≈
⎟⎟⎠
⎞⎜⎜⎝
⎛+∆−≈⎟⎟
⎠
⎞⎜⎜⎝
⎛+∆−
∑
∑
∑
0*
22
crist0
2
crist0
2
crist0
2
2
2
22
et on revient en sens inverse:
( ) ( )( )
( )( )
( )
( )
( )
( ) ( )rFEm
ru
eFEm
ru
eEm
Fru
eEmkFru
rueEmkF
rueEmkFrV
m
rki
kk
rki
kk
rki
kk
rki
kk
krki
kk
rhr
hr
hr
hr
rh
rhrh
rr
rr
rr
rr
rr
rr
rr
rr
rrr
rr
⎟⎟⎠
⎞⎜⎜⎝
⎛+∆−=
⎟⎟⎠
⎞⎜⎜⎝
⎛+∆−=
⎟⎟⎠
⎞⎜⎜⎝
⎛+∆−=
⎟⎟⎠
⎞⎜⎜⎝
⎛+=
⎟⎟⎠
⎞⎜⎜⎝
⎛+≈
⎟⎟⎠
⎞⎜⎜⎝
⎛+≈⎟⎟
⎠
⎞⎜⎜⎝
⎛+∆−
∑
∑
∑
∑
∑
0*
2
0
.0*
2
0
.0*
2
0
.0*
22
0
0.
0*
22
.0*
22
crist0
2
2
2
2
2
2
22ϕ
L'équation de Schrödinger complète que l'on doit résoudre s'écrit maintenant
( ) ( ) ( ) ( ) ( ) ( ) ( )rFruErFrurVrFEm
ru rrrrrrhr000*
2
0 2=+⎟⎟
⎠
⎞⎜⎜⎝
⎛+∆− ,
ou encore, après suppression du facteur commun u0 et réorganisation des termes,
( ) ( ) ( ) ( )rFEErFrVm
rrrh0*
2
2−=⎟⎟
⎠
⎞⎜⎜⎝
⎛+∆− .
En résumé, l'effet du cristal n'est plus représenté par le potentiel cristallin et le terme périodique u0 mais par la courbure de bande et le zéro d'énergie. Ce qu'il faut retenir, c'est qu'il reste à résoudre une pseudo-équation de Schrödinger où:
- l'énergie "cinétique" fait appel à la masse effective, - la fonction propre est la fonction enveloppe, - l'origine des énergies est le bas de la bande.
La démarche consiste alors à résoudre cette équation, puis à vérifier la seule hypothèse, qui est que la fonction kFr ne prend de valeurs notables qu'au voisinage de k=0, c'est-à-dire que la fonction enveloppe ainsi trouvée est peu localisée.

85
II. Transport
1. Transport diffusif et conductivité Ce paragraphe est un simple résumé de ce qui se passe dans un matériau massif: transport diffusif, permettant de définir une conductivité locale.
a. Rappel : équation de Boltzmann A l'équilibre, les porteurs de charge responsables de la conductivité électrique sont répartis sur les états des bandes partiellement occupées (électrons dans la bande de conduction d'un semiconducteur, trous dans la bande de valence d'un semiconducteur, électrons dans les bandes s et d d'un métal...), suivant la distribution de Fermi-Dirac
1exp
1)(+⎟⎟
⎠
⎞⎜⎜⎝
⎛ −=
TkE
Ef
B
µµ
Hors équilibre, par exemple en présence d'un champ électrique Er
, la répartition des porteurs sera décrite par une fonction de distribution ),,( tkrg
rr obéissant à l'équation de Boltzmann:
colldtdg
kgEq
rg
mk
tg
⎥⎦⎤
⎢⎣⎡=
∂∂
−∂∂
+∂∂
rh
r
r
rh ..* .
Cette équation peut se reconstruire à partir de
colldtdg
dtkd
kg
dtrd
rg
tg
⎥⎦⎤
⎢⎣⎡=
∂∂
+∂∂
+∂∂
r
rr
r ..
en remplaçant les gradients par leurs expressions dans les équations du mouvement d'un paquet d'onde, en particulier pour des électrons occupant une bande parabolique et soumis à un champ électrique:
h
r
h
rrrh
rh
rr EqF
dtkd
mk
kE
dtrd
k−
===∂∂
== ,1*v .
Le dernier terme de l'équation de Boltzmann ( )colldtdg / représente toute une série de "termes de collisions" qui perturbent le mouvement des porteurs, sur lesquels on reviendra plus loin. On s'attend à ce que l'équation de Boltzmann ramène la fonction de distribution hors équilibre
),,( tkrgrr vers la fonction à l'équilibre )(Ef µ .
Si l'on sait calculer la fonction g, on peut en déduire la densité de courant
( ) *,,mktkrgqj
k
rhrrr
r∑−= .
En réalité, puisque le courant est nul à l'équilibre (c'est-à-dire si µfg = ), il suffit de connaître
( )µfg − et de calculer ( ) ( )[ ] *)(,,m
kkEftkrgqjk
rhrrr
r∑ −−= µ . En particulier, le retour à
l'équilibre (de la fonction g vers la distribution de Fermi f) peut parfois se décrire par

86
l'approximation du temps de relaxation, τ
µfgdtdg
coll
−−=⎥⎦
⎤⎢⎣⎡ ; alors on calculera le courant en
régime permanent proche de l'équilibre sous la forme
[ ] ( )∑∑∑ ⎥⎦
⎤⎢⎣
⎡
∂∂−
+∂∂
=⎥⎦⎤
⎢⎣⎡=−−=
kk collk mk
kgEq
rg
mkq
mk
dtdgq
mkfgqj
rrr
rh
rh
r
r
rh
rh
rhr
**** ..ττµ
Dans cette dernière expression, le premier terme est dû à une distribution des porteurs inhomogène dans l'espace et donc correspond au courant de diffusion; le second terme est provoqué directement par le champ appliqué et correspond donc au courant de dérive. Des manipulations plus ou moins évidentes et plus ou moins rigoureuses permettent de les mettre sous la forme usuelle (celle du modèle de Drude)
rnqDjdif r
r
∂∂
= et Em
nqEqnEjdrv
rrrr*
2 τασ === .
On a ainsi défini des quantités locales: la mobilité α et la conductivité σ, ainsi que le coefficient de diffusion D. Pour définir ces quantités locales, on a utilisé l'équation de Boltzmann, c'est-à-dire une équation différentielle, locale, qui décrit l'évolution d'une fonction de distribution, ),,( tkrg
rr . Ceci suppose que dans tout volume (de l'espace réel et de l'espace réciproque) et pour tout intervalle de temps, un grand nombre d'événements (les "collisions") se produisent, qui permettent d'ignorer toute corrélation dans le mouvement. L'électron effectue alors un mouvement diffusif, allant d'un centre de diffusion à l'autre, et perdant toute mémoire au moment de la collision. C'est cette condition qui va disparaître dans le cas des nanostructures. On peut ré-écrire l’expression du courant dans l’hypothèse d’un équilibre local, avec un potentiel chimique local ( )rrµ , ce qui conduit à écrire :
( ) 1)(exp
1),,(+⎟⎟
⎠
⎞⎜⎜⎝
⎛ −=
TkrkE
tkrg
B
rrrr
µ.
En explicitant l’expression du courant on est alors amené à définir le potentiel électro-chimique Φ−= qµµ~ , où Φ est le potentiel électrostatique. Le courant total (diffusion et dérive) est alors proportionnel au gradient de ce potentiel électrochimique. Il faut aussi noter que le courant de dérive fait intervenir la dérivée kg
r∂∂ . Pour calculer ce
terme, on peut utiliser µfg ≈ , donc ( ) *
2
1m
kffTkkE
Ef
kg
B
rh
rr µµ −−=∂∂
∂∂
≈∂∂ . On peut alors
chercher à relier la mobilité α au coefficient de diffusion. Des hypothèses simplificatrices permettent d'écrire la relation:
( ) ( )
( )∫∫=
dEdEdf
E
dEEfEqDµ
µ
ρ
ρ
α.
Pour un semiconducteur non dégénéré, la statistique de Fermi-Dirac à haute température ( µ−>> ETkB ) est semblable à la statistique classique de Maxwell-Boltzmann. La distribution µf prend des valeurs entre 0 et 1 pour tous les états concernés, et tous contribuent à la conduction. La relation conduit alors à la forme classique de la relation d'Einstein

87
TkqDB=
α.
Pour un métal ou un semiconducteur dégénéré, cette quantité ne prend des valeurs non-nulles que dans un intervalle étroit ( TkB≈ ) autour de FE=µ . Ce sont donc les états autour de FE qui jouent un rôle dans la conductivité. On montre que la relation d'Einstein devient alors
( )FEnqD
ρα= .
b. Les différents types de collisions Le terme ( )colldtdg / recouvre de nombreux mécanismes d'interactions avec des défauts (impuretés chargées ou neutres, défauts étendus, interfaces; phonons; autres porteurs; impuretés magnétiques, fluctuations magnétiques; etc...). Chacun de ces mécanismes réclame un traitement complet. Certains d'entre eux peuvent être représentés, dans l'équation de Boltzmann, par un temps de collision (ou plus exactement temps moyen entre deux collisions)
kτ ou )(Eτ , ce qui permet d'écrire k
fgdtdg
τ−
−= .
Les probabilités de collisions s'ajoutent dans ( )colldtdg / : cela signifie que ce sont les inverses des mobilités qui s'ajoutent, et que dans le cas où on peut définir un temps de collision, ce sont les inverses des temps qui s'ajoutent. Parmi ces mécanismes de "collisions", on distingue généralement deux catégories: - les collisions élastiques, qui changent le vecteur d'onde k
r à énergie constante: pour une
bande parabolique isotrope, seule la direction de kr
est donc modifiée. L'énergie étant conservée, la phase de la fonction d'onde peut donc être conservée. Ces mécanismes comprennent les interactions avec les impuretés (neutres ou chargées), avec des défauts ponctuels ou étendus, avec les défauts d'interfaces dans un puits quantiques (rugosité),... - les collisions inélastiques, qui changent l'énergie du porteur. La différence d'énergie est absorbée ou fournie par une autre "particule". La différence importante avec la classe précédente des mécanismes élastiques est que la phase de la fonction d'onde du porteur est alors perdue. Le temps entre deux mécanismes de collisions inélastique définit donc le temps de cohérence. Les "particules" intervenant dans les collisions inélastiques sont les phonons (acoustiques, optiques), les autres porteurs, les magnons... Les interactions entre porteurs jouent un rôle faible dans un semiconducteur peu dopé; on pourraient s'attendre à ce qu'elles jouent un rôle important dans un semiconducteur très dopé ou un métal; cependant des effets d'écrantage ont lieu et diminuent leur influence ( les interactions entre porteurs sont prises en compte dans la théorie du liquide de Fermi, qui conduit à remplacer les porteurs en interaction par des quasi-particules). Pour obtenir un temps de cohérence long, il reste à modérer l'effet des phonons, en général en diminuant la température... Ainsi, la figure 1 donne un exemple de mobilité dans un semiconducteur (courbe notée "bulk GaAs"). Des modèles détaillés permettent de remonter à l'origine d'une telle courbe, et montrent que:
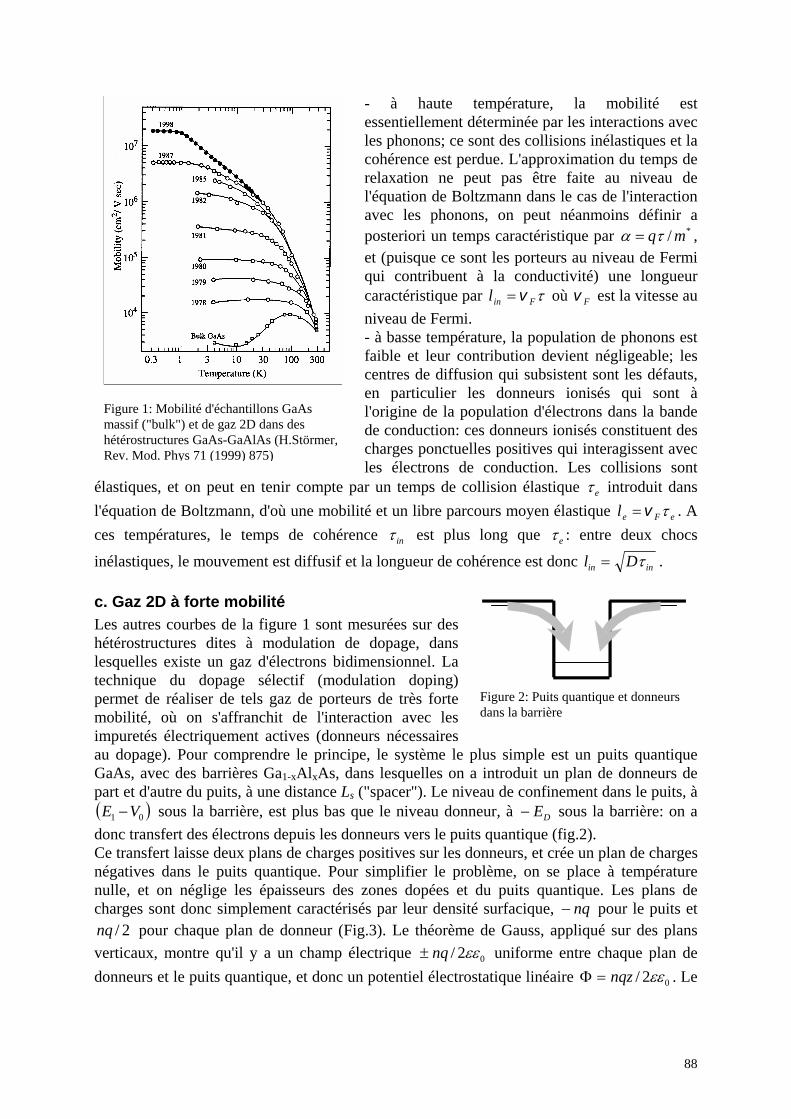
88
- à haute température, la mobilité est essentiellement déterminée par les interactions avec les phonons; ce sont des collisions inélastiques et la cohérence est perdue. L'approximation du temps de relaxation ne peut pas être faite au niveau de l'équation de Boltzmann dans le cas de l'interaction avec les phonons, on peut néanmoins définir a posteriori un temps caractéristique par */ mqτα = , et (puisque ce sont les porteurs au niveau de Fermi qui contribuent à la conductivité) une longueur caractéristique par τFinl v= où Fv est la vitesse au niveau de Fermi. - à basse température, la population de phonons est faible et leur contribution devient négligeable; les centres de diffusion qui subsistent sont les défauts, en particulier les donneurs ionisés qui sont à l'origine de la population d'électrons dans la bande de conduction: ces donneurs ionisés constituent des charges ponctuelles positives qui interagissent avec les électrons de conduction. Les collisions sont
élastiques, et on peut en tenir compte par un temps de collision élastique eτ introduit dans l'équation de Boltzmann, d'où une mobilité et un libre parcours moyen élastique eFel τv= . A ces températures, le temps de cohérence inτ est plus long que eτ : entre deux chocs
inélastiques, le mouvement est diffusif et la longueur de cohérence est donc inin Dl τ= .
c. Gaz 2D à forte mobilité Les autres courbes de la figure 1 sont mesurées sur des hétérostructures dites à modulation de dopage, dans lesquelles existe un gaz d'électrons bidimensionnel. La technique du dopage sélectif (modulation doping) permet de réaliser de tels gaz de porteurs de très forte mobilité, où on s'affranchit de l'interaction avec les impuretés électriquement actives (donneurs nécessaires au dopage). Pour comprendre le principe, le système le plus simple est un puits quantique GaAs, avec des barrières Ga1-xAlxAs, dans lesquelles on a introduit un plan de donneurs de part et d'autre du puits, à une distance Ls ("spacer"). Le niveau de confinement dans le puits, à ( )01 VE − sous la barrière, est plus bas que le niveau donneur, à DE− sous la barrière: on a donc transfert des électrons depuis les donneurs vers le puits quantique (fig.2). Ce transfert laisse deux plans de charges positives sur les donneurs, et crée un plan de charges négatives dans le puits quantique. Pour simplifier le problème, on se place à température nulle, et on néglige les épaisseurs des zones dopées et du puits quantique. Les plans de charges sont donc simplement caractérisés par leur densité surfacique, nq− pour le puits et
2/nq pour chaque plan de donneur (Fig.3). Le théorème de Gauss, appliqué sur des plans verticaux, montre qu'il y a un champ électrique 02/ εεnq± uniforme entre chaque plan de donneurs et le puits quantique, et donc un potentiel électrostatique linéaire 02/ εεnqz=Φ . Le
Figure 2: Puits quantique et donneurs dans la barrière
Figure 1: Mobilité d'échantillons GaAs massif ("bulk") et de gaz 2D dans des hétérostructures GaAs-GaAlAs (H.Störmer, Rev. Mod. Phys 71 (1999) 875)

89
potentiel vu par les électrons de charge –q est donc la somme du potentiel du puits et de l'énergie potentielle électrostatique,
)()( zqzV Φ− pour
sLz <<0 . A température nulle, si la densité de donneurs est supérieure à n/2, des donneurs neutres et ionisés coexistent dans chaque plan, et le niveau de Fermi passe donc au voisinage du niveau donneur. Par ailleurs, la densité d'électrons dans le puits quantique est obtenue en multipliant la distance
entre le niveau de Fermi et le bas du continuum 2D, par la densité d'états 2D 2*2 / hπρ mD = .
Le bilan s'établit en suivant les potentiels tout le long de la structure et en écrivant que le niveau de Fermi est uniforme:
D
SD
nEVLnqE2
100
2
2 ρεε++−=− ,
d'où
⎟⎟⎠
⎞⎜⎜⎝
⎛+
−−=
*
2
0
210
2 mLq
EEVnS
D
hπεε
Cette analyse est très simplifiée car le profil de potentiel, les énergies de confinement et les transferts de charges doivent être calculés de façon auto-cohérente. Cependant il met bien en évidence deux paramètres essentiels du transfert: la profondeur du puits, et la largeur de la zone d'espacement (spacer). En pratique, on utilise une simple hétérojonction GaAs-GaAlAs, ou Si-SiO2, le transfert de charge assurant le potentiel de façon auto-consistante (figure 4). De plus, des contacts électriques (grille, ou gate) sont déposés à la surface, et permettent de moduler la densité de porteurs
en appliquant un champ électrique (voir exercice). En outre, on peut grâce à ces contacts moduler latéralement le gaz de porteurs, et créer des fils, des boîtes, ou des profils plus compliqués..., qui sont très utilisés pour les études de transport dans des structures confinées (Fig. 5).
Fig. 4: Principe d'un échantillon à gaz 2D basé sur l'interface Si-SiO2 (a) ou GaAs-GaAlAs (b), et schéma de la bande de conduction (c) (H.Störmer, Rev. Mod. Phys 71
Densité de charges
Champ électrique
Potentiel électrostatique
Potentiel total
EF
Donneurs
Puits quantique
Figure 3: charges, champ, potentiel électrostatique et potentiel total dans un puits quantique à dopage sélectif.

90
Application: gaz de très haute mobilité. Comme les porteurs sont maintenant séparés spatialement des donneurs ionisés (fig.4c), la mobilité à basse température devient très forte. On en verra des applications plus loin, mais toutes les études autour de l'effet Hall entier ou fractionnaire sont faites sur de telles structures. De plus, le gain de mobilité est significatif même à température ambiante, ce qui permet la fabrication des HEMT (High Electron Mobility Transistors) qui équipent les circuits haute fréquence. La concurrence s'est développée entre de telles structures à base d'hétérostructures GaAs/GaAlAs, ou à base de Ge-Si, en particulier pour l'équipement des téléphones portables. On pourra calculer, en supposant
-212 cm10≈n , τe, le,et eFlk pour l'échantillon 1998 de la figure 1, et commenter cette dernière quantité, en particulier pour les données de 1978.
2. Systèmes 1D ; quantification de la conductance
On s'intéresse ici à un conducteur de
dimensions petites par rapport à el et inl , joignant deux réservoirs d'électrons entre lesquels on applique une faible tension. De nombreux résultats dans ce domaine sont obtenus à partir de gaz 2D, modulés latéralement par des contacts électriques (Fig. 5 et 6) ou par attaque chimique. La structure de bande dans les semiconducteurs permet de ne peupler qu'un certain
nombre d'états confinés pour des dimensions de confinement raisonnables. Les contacts métalliques permettent aussi d'ajuster la population du gaz d'électrons et donc le nombre d'états occupés. Les dimensions utiles sont plus difficiles à atteindre dans le cas des métaux. On considère donc deux réservoirs de charges, avec une différence de potentiel V, reliés par un fil conducteur de longueur L et de largeur w (fig.6 et 7). Dans la description classique, le gaz bidimensionnel de ce fil est décrit en tout point par une conductivité locale σ (quelle est sa dimension?), et le courant dans le canal est
LwVI σ
= . On définit donc une conductance du fil, qui est une fonction d'une part de la
géométrie du fil, d'autre part d'une propriété locale (la conductivité) uniforme. D'après ce qui
Fig. 6: Fil 1D réalisés par contacts métalliques sur un gaz d'électrons 2D (Pasquier, thèse Paris 1994)
Fig. 5: Contacts en surface d'un échantillon GaAs-GaAlAs à gaz d'électrons 2D. Les contacts sont en clair, la surface du GaAs en sombre. Lorsque aucun potentiel n'est appliqué, on a un gaz 2D. Lorsqu'un même potentiel négatif est appliqué aux deux contacts marqués (a), ils définissent entre eux une zone 1D "fil quantique". Il en va de même des contacts (c). Lorsqu'un potentiel plus fort est appliqué aux contacts (a, c), les zones 1D sont coupées et cela permet d'isoler une zone 0D "boîte quantique" dont la dimension peut être ajuster par les contacts (b). (D.Pasquier, thèse Paris 1994)

91
précède, l'existence d'une telle quantité suppose que les dimensions du fil (L et w) soient grandes devant el et inl .
L'extrême inverse est le cas où L et w sont petits devant el et inl . Alors un électron va passer d'un réservoir à l'autre en occupant un état propre du système quantique que constitue l'ensemble du fil (fig.7). Les fonctions enveloppes dans le fil s'écrivent
),()exp( yxLikz ψ
où z est la direction du fil, et ),( yxψ décrit le confinement
suivant les directions perpendiculaires au fil. Chaque état contribue à la densité de courant par 2
* ),(1 yxLm
kqjk ψh−= .
Le courant total est obtenu en sommant les contributions de chaque état occupé dans le fil:
kLm
qdxdyyxjJ
JJ
kk
kk
*),( h−==
=
∫∫
∑
Quels sont les états à prendre en compte? Chaque extrémité du fil est reliée à un "réservoir", qui accepte les électrons arrivant du fil (ceux-ci sont diffusés dans le réservoir avec un libre parcours moyen el et thermalisés au bout d'une longueur inl , et les dimensions du réservoir sont grandes devant ces deux longueurs caractéristiques) et fournit au fil des électrons avec une population définie par le potentiel chimique du réservoir ( 1µ ou 2µ avec
qV−=− 21 µµ ). La population des états de 0>k du fil est donc décrite par la distribution de Fermi de potentiel chimique 1µ , et celle des états de 0<k par la distribution de Fermi de potentiel chimique 2µ . Le courant total devient alors
( )
( )∫
∑
∑∑
∞
>
<>
−−
=
−−
=
⎥⎦
⎤⎢⎣
⎡+
−=
021*
021*
02
01*
2
)()(
dkffkLLm
q
ffkLm
q
EkfEkfLm
qI
k
kk
µµ
µµ
µµ
πh
h
h
d'où, en utilisant *2 / mdkkdE h= , et la relation ( ) ( ) qVdEff −=−=−∫ 2121 µµµµ ,
valable à basse température,
VhqI
2
=
x
z
µ1 µ2
Fig. 7: Parcours d'un électron: mouvement diffusif dans le réservoir 1, transmission cohérente via un canal dans le fil, diffusion et thermalisation dans le réservoir 2.
Fig.8: représentation schématique d'un contact ponctuel par une pointe STM 'Agraït et al., con-mat/0208239
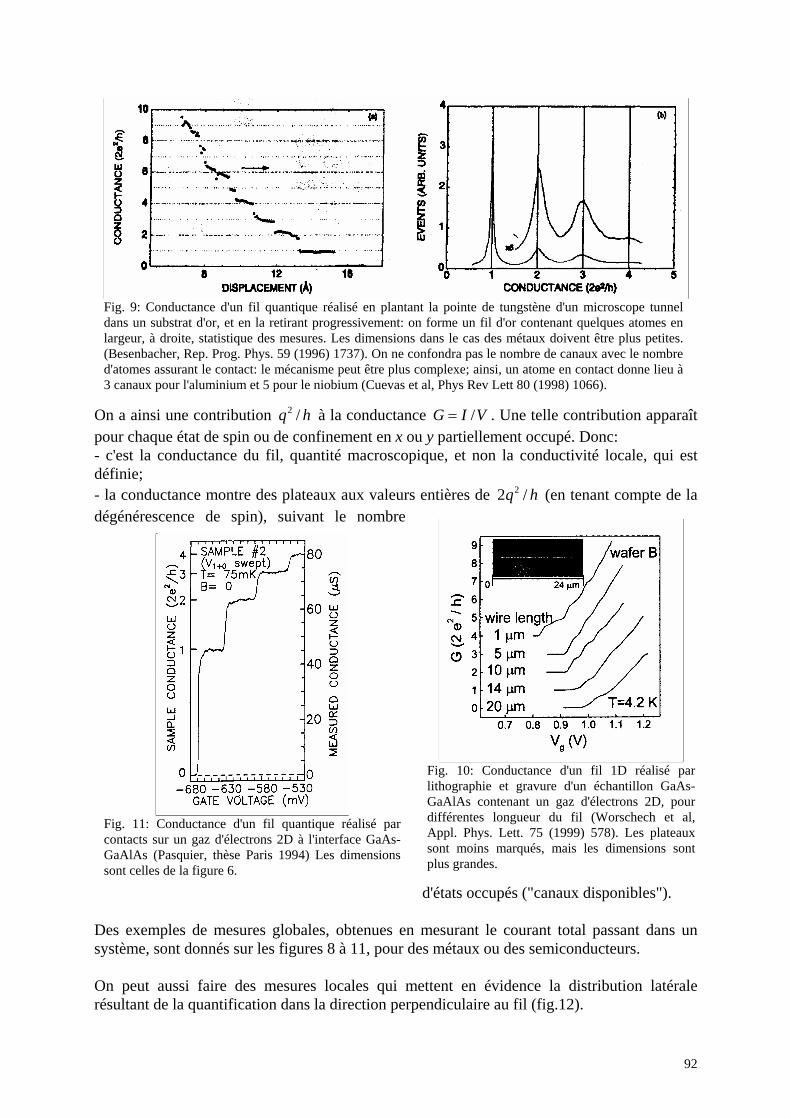
92
On a ainsi une contribution hq /2 à la conductance VIG /= . Une telle contribution apparaît pour chaque état de spin ou de confinement en x ou y partiellement occupé. Donc: - c'est la conductance du fil, quantité macroscopique, et non la conductivité locale, qui est définie; - la conductance montre des plateaux aux valeurs entières de hq /2 2 (en tenant compte de la dégénérescence de spin), suivant le nombre
d'états occupés ("canaux disponibles"). Des exemples de mesures globales, obtenues en mesurant le courant total passant dans un système, sont donnés sur les figures 8 à 11, pour des métaux ou des semiconducteurs. On peut aussi faire des mesures locales qui mettent en évidence la distribution latérale résultant de la quantification dans la direction perpendiculaire au fil (fig.12).
Fig. 9: Conductance d'un fil quantique réalisé en plantant la pointe de tungstène d'un microscope tunnel dans un substrat d'or, et en la retirant progressivement: on forme un fil d'or contenant quelques atomes en largeur, à droite, statistique des mesures. Les dimensions dans le cas des métaux doivent être plus petites. (Besenbacher, Rep. Prog. Phys. 59 (1996) 1737). On ne confondra pas le nombre de canaux avec le nombre d'atomes assurant le contact: le mécanisme peut être plus complexe; ainsi, un atome en contact donne lieu à 3 canaux pour l'aluminium et 5 pour le niobium (Cuevas et al, Phys Rev Lett 80 (1998) 1066).
Fig. 10: Conductance d'un fil 1D réalisé par lithographie et gravure d'un échantillon GaAs-GaAlAs contenant un gaz d'électrons 2D, pour différentes longueur du fil (Worschech et al, Appl. Phys. Lett. 75 (1999) 578). Les plateaux sont moins marqués, mais les dimensions sont plus grandes.
Fig. 11: Conductance d'un fil quantique réalisé par contacts sur un gaz d'électrons 2D à l'interface GaAs-GaAlAs (Pasquier, thèse Paris 1994) Les dimensions sont celles de la figure 6.
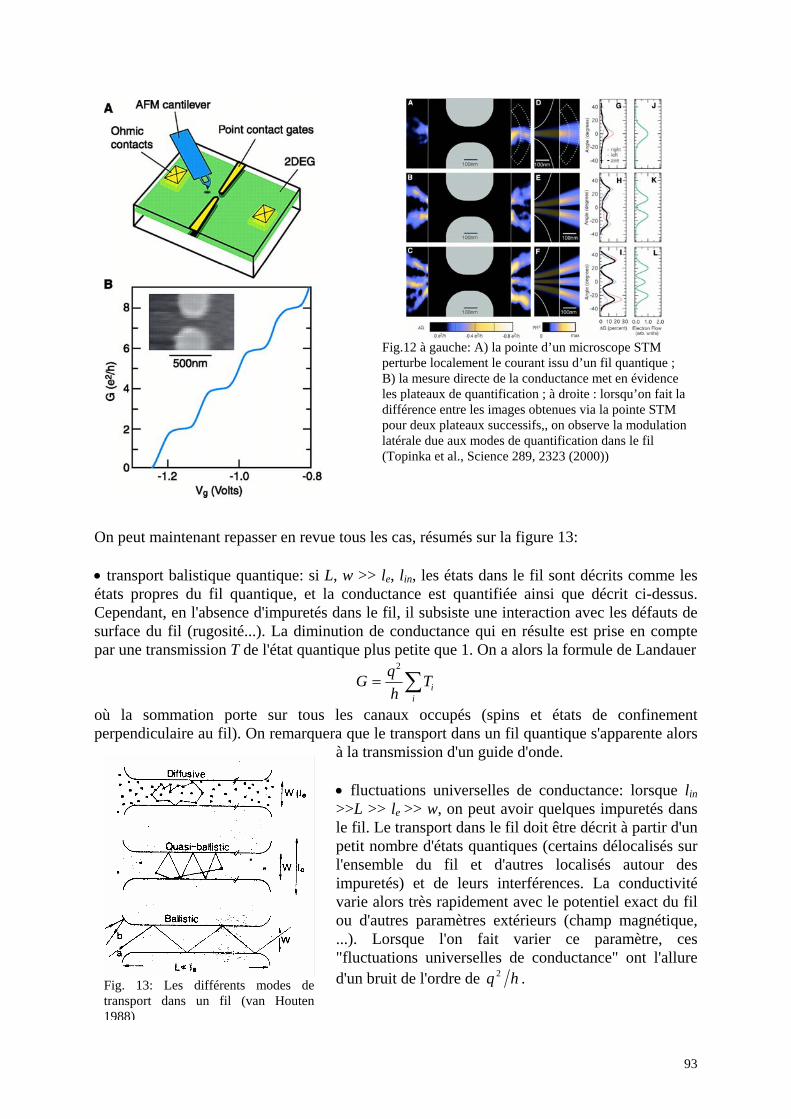
93
On peut maintenant repasser en revue tous les cas, résumés sur la figure 13: • transport balistique quantique: si L, w >> le, lin, les états dans le fil sont décrits comme les états propres du fil quantique, et la conductance est quantifiée ainsi que décrit ci-dessus. Cependant, en l'absence d'impuretés dans le fil, il subsiste une interaction avec les défauts de surface du fil (rugosité...). La diminution de conductance qui en résulte est prise en compte par une transmission T de l'état quantique plus petite que 1. On a alors la formule de Landauer
∑=i
iThqG
2
où la sommation porte sur tous les canaux occupés (spins et états de confinement perpendiculaire au fil). On remarquera que le transport dans un fil quantique s'apparente alors
à la transmission d'un guide d'onde. • fluctuations universelles de conductance: lorsque lin >>L >> le >> w, on peut avoir quelques impuretés dans le fil. Le transport dans le fil doit être décrit à partir d'un petit nombre d'états quantiques (certains délocalisés sur l'ensemble du fil et d'autres localisés autour des impuretés) et de leurs interférences. La conductivité varie alors très rapidement avec le potentiel exact du fil ou d'autres paramètres extérieurs (champ magnétique, ...). Lorsque l'on fait varier ce paramètre, ces "fluctuations universelles de conductance" ont l'allure d'un bruit de l'ordre de hq 2 .
Fig.12 à gauche: A) la pointe d’un microscope STM perturbe localement le courant issu d’un fil quantique ; B) la mesure directe de la conductance met en évidence les plateaux de quantification ; à droite : lorsqu’on fait la différence entre les images obtenues via la pointe STM pour deux plateaux successifs,, on observe la modulation latérale due aux modes de quantification dans le fil (Topinka et al., Science 289, 2323 (2000))
Fig. 13: Les différents modes de transport dans un fil (van Houten 1988)

94
• localisation faible: lorsque lin >>L >> w>> le, les états sont localisés. A température nulle, la conductivité est nulle (aucun état n'est délocalisé sur toute la longueur du fil). Une conductivité par saut entre états localisés apparaît si l'on augmente la température, ce qui permet de compenser les faibles différences d'énergie entre états localisés grâce aux phonons. On finit par retrouver le cas du transport diffusif (équation de Boltzmann) lorsque L >> w>> lin . Tous ces régimes donnent lieu à un grand nombre d'études de "physique mésoscopique". Lorsque la conductance à travers un fil unique est quantifiée, on observe des interférences lorsque plusieurs chemins sont possibles, voire du chaos lorsque la géométrie devient plus complexe.
3. Effet tunnel On s'intéresse maintenant au transport de courant entre deux conducteurs séparés par une barrière.
a. Fonctions d'onde en présence d'une barrière tunnel rectangulaire
On considère une barrière de hauteur V0 et de largeur L. Les origines de position (axe z perpendiculaire à la barrière) et d'énergie sont choisies au milieu de la barrière et au bas de la bande la plus basse. Comme dans le cas du puits quantique, on va utiliser le modèle de la fonction enveloppe, en transposant les résultats du problème à une dimension z des cours de mécanique quantique.
On doit trouver les solutions de l' équation de Schrödinger
)()()()(2 2
22
zEzzVzzm
ϕϕϕ =+∂∂
−h ,
avec 0)( =zV pour 2/Lz −< et 2/Lz > , et 0)( VzV = pour 2/2/ LzL <<− Pour une énergie E<V0, la fonction s'écrit
ikzikz eBeAz −+=)(ϕ pour 2/Lz −< zkzk eDeC '' −+= pour 2/2/ LzL <<−
ikzikz eBeA −+= ~~ pour 2/Lz > où l'on a posé mkE 2/22h= et mkEV 2/'22
0 h=− .
La continuité de la fonction d'onde et de sa dérivée aux interfaces permet de calculer A~ et B~ en fonction de A et B. La relation est en général écrite sous forme matricielle
V0
-L/2 0 L/2 Fig. 14: Barrière de potentiel entre deux conducteurs.

95
( ) ⎥⎦
⎤⎢⎣
⎡=⎥
⎦
⎤⎢⎣
⎡BA
kMBA~~
où la matrice de transmission )(kM vaut, dans ce cas particulier d'une barrière rectangulaire
( ) ( ) ( )
( ) ( ) ( ) ⎥⎥⎥⎥
⎦
⎤
⎢⎢⎢⎢
⎣
⎡
⎥⎦
⎤⎢⎣
⎡ −−
+
+−⎥
⎦
⎤⎢⎣
⎡ −+
=
−
ikL
ikL
eLkkk
kkiLkLkkk
kki
Lkkk
kkieLkkk
kkiLkkM
'sinh'2''cosh'sinh
'2'
'sinh'2''sinh
'2''cosh
)( 2222
2222
.
On peut calculer le courant de probabilité associé à la fonction d'onde:
[ ] ⎥⎦⎤
⎢⎣⎡ −=−=⎥⎦
⎤⎢⎣⎡ −−=
2222 ~~21)( BA
mkBA
mk
dzd
dzd
mizj hhh ϕϕϕϕ .
Le courant de probabilité d'une fonction propre est indépendant de la position. Une telle matrice de transmission peut être définie pour le cas général d'un barrière de forme
)(zV quelconque. La symétrie dans le changement kk −→ , et la conservation du courant, impliquent que
AABB MM = , BAAB MM = et 1)( =MDet , d'où aussi ⎥⎦
⎤⎢⎣
⎡−
−=−
AABA
BAAA
MMMM
M 1
b. Transparence de la barrière On peut calculer les coefficients de transmission et de réflexion de la barrière en cherchant une solution telle que 0~ =B et 1~
=A . On trouve AAMA = et BAMB −= , d'où le coefficient de
réflexion ( ) 2AABA MMkR = et le coefficient de transmission ( ) 21 AAMkT = .
En particulier, pour la barrière rectangulaire
( ) ( ) ( )
( ) ( )LkEVE
V
Lkkk
kkLkkk
kkLkT
'sinh4
1
'sinh'2'1'sinh
'2''cosh1
2
0
20
2222
2222
2
−+=
⎟⎟⎠
⎞⎜⎜⎝
⎛ ++=⎟⎟
⎠
⎞⎜⎜⎝
⎛ −+=
.
On retrouve 1=T pour une barrière transparente ( )0' →Lk , alors que pour une barrière haute et épaisse
( ) ( )
( ) hEVmk
LkEVE
VT
−=
−−
≈
0
0
20
2'
'2exp16.
On néglige souvent le préfacteur, qui varie lentement par rapport au terme exponentiel.
c. Barrière de forme quelconque Le calcul ci-dessus est possible pour une barrière rectangulaire, ce qui n'est que très rarement réalisé. On peut traiter le cas plus général d'une barrière de forme quelconque )(zV en utilisant l'approximation WKB: cette approximation montre que l'on peut remplacer le terme calculé ci-dessus par
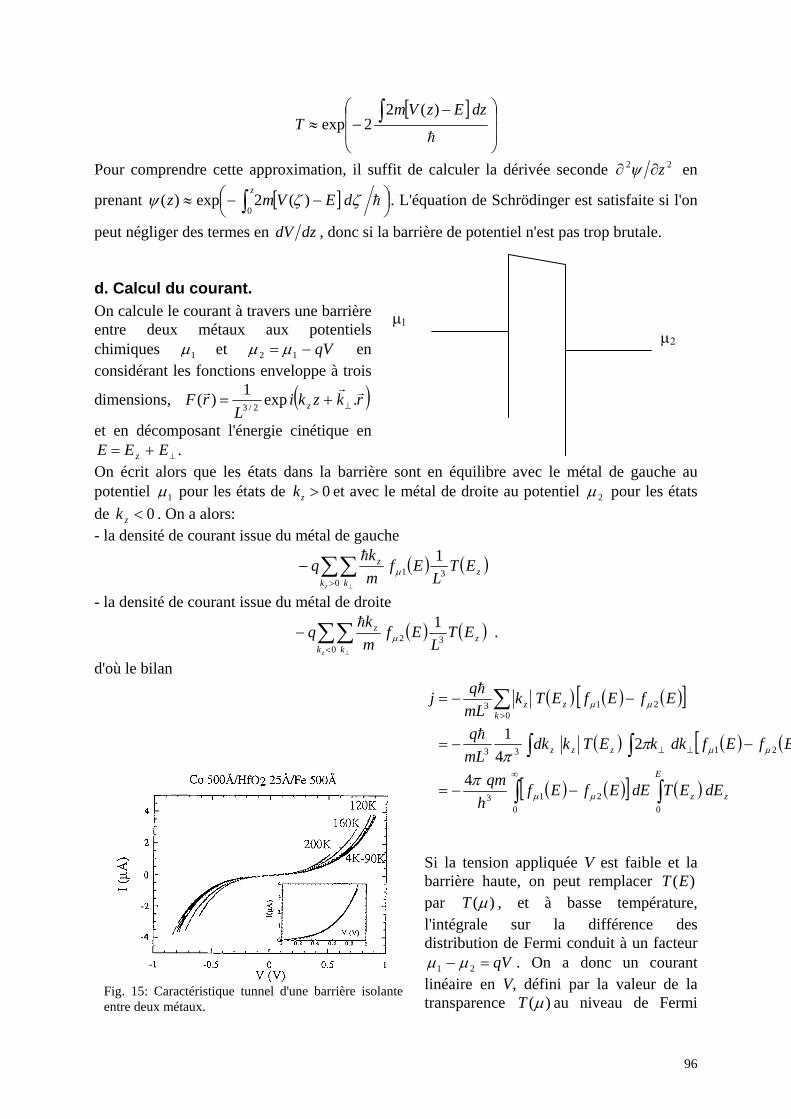
96
[ ]⎟⎟
⎠
⎞
⎜⎜
⎝
⎛ −−≈ ∫
h
dzEzVmT
)(22exp
Pour comprendre cette approximation, il suffit de calculer la dérivée seconde 22 z∂∂ ψ en
prenant [ ] ⎟⎠⎞⎜
⎝⎛ −−≈ ∫ hζζψ dEVmz
z
0)(2exp)( . L'équation de Schrödinger est satisfaite si l'on
peut négliger des termes en dzdV , donc si la barrière de potentiel n'est pas trop brutale.
d. Calcul du courant. On calcule le courant à travers une barrière entre deux métaux aux potentiels chimiques 1µ et qV−= 12 µµ en considérant les fonctions enveloppe à trois
dimensions, ( )rkzkiL
rF zrrr .exp1)( 2/3 ⊥+=
et en décomposant l'énergie cinétique en .⊥+= EEE z
On écrit alors que les états dans la barrière sont en équilibre avec le métal de gauche au potentiel 1µ pour les états de 0>zk et avec le métal de droite au potentiel 2µ pour les états de 0<zk . On a alors: - la densité de courant issue du métal de gauche
( ) ( )∑∑> ⊥
−0
311
zk kz
z ETL
Efmkq µh
- la densité de courant issue du métal de droite
( ) ( )∑∑< ⊥
−0
321
zk kz
z ETL
Efmkq µh .
d'où le bilan
( ) ( ) ( )[ ]
( ) ( ) ([
( ) ( )[ ] ( ) z
E
z
zzz
zk
z
dEETdEEfEfh
qm
EfEfdkkETkdkmLq
EfEfETkmLqj
∫∫
∫∫
∑
∞
⊥⊥
>
−−=
−−=
−−=
00213
2133
210
3
4
24
1
µµ
µµ
µµ
π
ππ
h
h
Si la tension appliquée V est faible et la barrière haute, on peut remplacer )(ET par )(µT , et à basse température, l'intégrale sur la différence des distribution de Fermi conduit à un facteur
qV=− 21 µµ . On a donc un courant linéaire en V, défini par la valeur de la transparence )(µT au niveau de Fermi
µ1 µ2
Fig. 15: Caractéristique tunnel d'une barrière isolante entre deux métaux.

97
pour une barrière rectangulaire. Si V augmente la barrière devient trapézoïdale et lorsque 0VV ≈ la transparence augmente, et le courant également. On a donc une caractéristique
)(VI surlinéaire (fig.15).
e. Calcul de perturbation et densité d'états Le calcul ci-dessus, de type masse effective, est souvent impossible à mettre en place si l'on considère une barrière isolante entre deux métaux de structures de bandes différentes . On utilise alors une formule qui ressemble à la règle d'or de Fermi, et qui s'écrit comme somme de deux courants opposés. La densité de courant de gauche à droite s'écrit
( ) ( ) ( )[ ]dgdgg d
dg EfEfEETqj 21, 12µµδπ
−−−= ∑∑h
a
où les sommations portent sur les états de gauche ( )g occupés, d'énergie Eg, et les états de droites ( )d vides, ainsi que le précisent les distributions de Fermi if µ correspondant aux potentiels chimiques iµ . On ne garde que des transferts élastiques. Si on peut considérer que
dgT , ne dépend que de l'énergie E (ce qui suppose qu'on n'a plus la conservation du vecteur d'onde comme c'était le cas ci-dessus: cette conservation était liée à l'utilisation d'un modèle simple du style fonction enveloppe), en remplaçant chaque somme discrète par une intégrale continue sur la densité d'états, et en utilisant la conservation d'énergie imposée par la fonction δ, on obtient une expression
( ) ( )[ ] dEEfEfETEEAj dg 21)()()( µµρρ −= ∫ Le courant tunnel est ainsi décrit à partir de la densité d'états dans le matériaux de gauche, du terme tunnel, et de la densité d'états dans le matériau de droite. Cela signifie que le terme de transfert, ou plutôt )(ETA , doit tenir compte de toute la physique microscopique (nature des états électroniques dans les trois matériaux et leur mélange aux interfaces), et que son calcul
n'est pas simple: on se contente souvent de prendre [ ]
⎟⎟
⎠
⎞
⎜⎜
⎝
⎛ −−≈ ∫
h
dzEzVmT
)(22exp et de
considérer A comme un paramètre ajustable... Références complémentaires: Calcul de base: J.G.Simmons, J. Appl. Phys. 34 (1963) 1793; R.Stratton, J. Phys. Chem. Solids 23 (1962) 1177 Discussion de fond: Hauge et Stovneng, Rev. Mod. Phys. 61 (1989) 917
4. L'approche "bottom-up": exemples en électronique moléculaire Les exemples précédents étaient issus de l'approche "top-down": on part d'un échantillon 2D (tel qu'un puits quantique ou une hétérojonction de semiconducteurs ou encore une couche métallique), ou même d'un échantillon 3D; on en réduit ensuite les dimensions, par gravure ou par formation d'un confinement latéral). Une approche opposée est dite "bottom-up": on s'appuie sur l'auto-assemblage d'une structure 0D (boîte quantique, plots métalliques) ou sur l'autoassemblage sous forme d'un réseau de structures 0D: on en a vu des exemples au chapitre 5 de la première partie et au premier chapitre de la seconde partie. Pour les mesures de transport, de nombreuses études sont développées actuellement sur des

98
structures issues de la chimie moléculaire. L'exemple le plus avancé est celui des nanotubes de carbone. Un autre domaine s'appuie sur des molécules organiques contactées dans des structures hybrides.
a. Du graphite aux nanotubes Le carbone, sous sa forme massive, peut présenter deux structures cristallines différentes, qui ont des propriétés physiques très différentes. Le diamant a une structure de type ..."diamant" (la même que le silicium). Chaque atome est en hybridation sp3, et il est entouré d'un tétraèdre régulier d'autres atomes de carbone. Le diamant est un semiconducteur cubique (avec des propriétés générales relativement isotropes), à grande bande interdite indirecte. Le graphite a une structure en feuillets. Chaque atome est en configuration sp2, et les atomes forment un plan de structure hexagonale: le plan de graphène (fig.16). La maille élémentaire dans le plan est définie par les deux vecteurs
⎟⎟⎟
⎠
⎞
⎜⎜⎜
⎝
⎛ −=
⎟⎟⎟
⎠
⎞
⎜⎜⎜
⎝
⎛=
02321
,0
2321
21 aaaa rr ,⎟⎟⎟
⎠
⎞
⎜⎜⎜
⎝
⎛=
100
cr
où nm246.03 == da , d étant la longueur de la liaison. A partir de ces deux vecteurs de base dans le plan et d'un vecteur perpendiculaire, on déduit le réseau réciproque dans le plan (kx, ky):
( ) ⎟⎟⎟
⎠
⎞
⎜⎜⎜
⎝
⎛=
×=
031
12
,, 21
2*1 acaa
caa πrrr
rrr ,
⎟⎟⎟
⎠
⎞
⎜⎜⎜
⎝
⎛ −=
031
12*
2 aa πr
et la première zone de Brillouin (fig. 17). Les points de haute symétrie sont ( )0,0,0=Γ ,
( )0,34,0 aKP π−== , ( )0,0,34 aMQ π== . Dans le graphite, les atomes de deux plans adjacents ont une interaction de type Van der Waals. Les propriétés sont très anisotropes, avec une bonne conductivité dans les directions du plan. La structure de bande dans le plan est décrite sur la figure 18. Elle comprend des états π, dont la dispersion est décrite de façon approchée par
Fig. 16: Appenzeller et al., Carbon nanotubes for data processing; in Nanoelectronics and Information Technology, R.Waiser ed., Wiley
M
K Γ
Fig. 17: première zone de Brillouin du plan de graphène.
*1ar
*2ar

99
l'équation
( ) 200 2
cos42
cos2
3cos41,⎢⎢⎣
⎡⎜⎜⎝
⎛+⎟⎟
⎠
⎞⎜⎜⎝
⎛⎟⎟⎠
⎞⎜⎜⎝
⎛+±=
kakakEkkE yyxyx γ
Dans cette expression, γ0 représente l'intégrale de transfert entre plus proches voisins (dans une description de type liaisons fortes) et a=0.246 nm est le paramètre de maille dans le plan de graphène. Cette dispersion est formée de deux nappes symétriques par rapport au plan E=E0. Ces deux nappes se touchent en E= E0, pour les points K de la zone de
Brillouin. La bande inférieure est remplie à température nulle, la bande supérieure est vide. Le niveau de Fermi est donc entre les deux bandes à E= E0, à une énergie où la densité d'états est nulle sans qu'il y ait ouverture d'un gap: il s'agit d'un semi-métal (dans le graphite, les interactions de van der Waals causent un recouvrement de 40 meV des deux bandes et on a donc un métal). On a découvert au cours des dernières années des nanostructures aux propriétés très particulières. Tout d'abord, la série des fullerènes, avec comme prototype le C60. C'est un arrangement régulier d'hexagones et de pentagones de carbone, formant une molécule covalente géante. Ces objets ont des propriétés mécaniques et électroniques exceptionnelles, mais sont surtout des objets de laboratoire. On a ainsi pu réaliser des interférences de deux molécules de fullerènes (Arndt et al., Nature 401, 680 (1999)). Plus récemment, les nanotubes de carbone (S.Iijima, Nature 354, 56 [1991]) ont suscité un intérêt tout particulier, parce qu'ils permettent d'imaginer un grand nombre d'applications. Ces nanotubes correspondent à des rubans de graphène enroulés. Il existe des nanotubes mono-paroi (un seul ruban enroulé), multi-paroi (plusieurs tubes concentriques), des faisceaux de nanotubes... La figure 19 (S. Iijima et al, Nature
Fig. 18: Structure de bande du graphène. Noter le niveau de Fermi, qui se trouve entre les bandes π liantes et antiliantes (Painter and Ellis, Phys Rev B 1, 4747 (1970)); attention aux notations: Q=M, P=K.
Fig. 19: Iijima et al., Nature 408, 50

100
408, 50 (2000)) représente une image en microscopie en transmission, d'un nanotube multiparoi abritant un nanotube de 0.4 nm. Plusieurs méthodes d'élaboration sont mises au point actuellement: décharge électrique (dans un arc), évaporation laser, CVD (chemical vapor deposition), recristallisation de fullerènes... On essaie de trier les nanotubes mono-paroi, de les accrocher sur des plots (fig. 20, croissance catalytique sur des plots de cobalt; ou sur des pointes STM), de les doper, de les fonctionnaliser...
b. Propriétés électroniques Le motif de base du plan de graphène est un hexagone d'atomes de carbone. Pour décrire un nanotube (fig. 21), on va isoler un ruban de largeur C, taillé dans le plan de graphène, et
Fig. 20: Nanotubes de carbone reliant des plots de cobalt (AM Bonnot, LEPES, V.Bouchiat et T.Fournier, CRTBT)
Fig. 21: Exemples de nanotubes de carbone, avec plusieurs types de vecteur C; Appenzeller et al., Carbon nanotubes for data processing; in Nanoelectronics and Information Technology, R.Waiser ed., Wiley
Fig. 22: Cas d'un nanotube "chaise" (3,3). L'axe du nanotube est suivant x, et toutes les valeurs de kx définissent la bande d'états 1D. Suivant ky, on a une valeur permise à ky = 0, passant par Γ, qui donne naissance à la bande à plus basse énergie pour les états liants et à plus haute énergie pour les états anti-liants. Les lignes permises aux valeurs suivantes, non nulles, de ky donnent naissance à deux bandes d'états liants doublement dégénérées (+ky et - ky ) de dispersion plus faible, et deux bandes d'états anti-liants. Enfin, la dernière valeur permise, en limite de zone de Brillouin, donne naissance à une dernière bande qui arrive au niveau de Fermi aux deux points K: on a donc une dispersion linéaire autour de ce point et une densité d'états finie au niveau de Fermi. (Appenzeller et al, op. cit.)
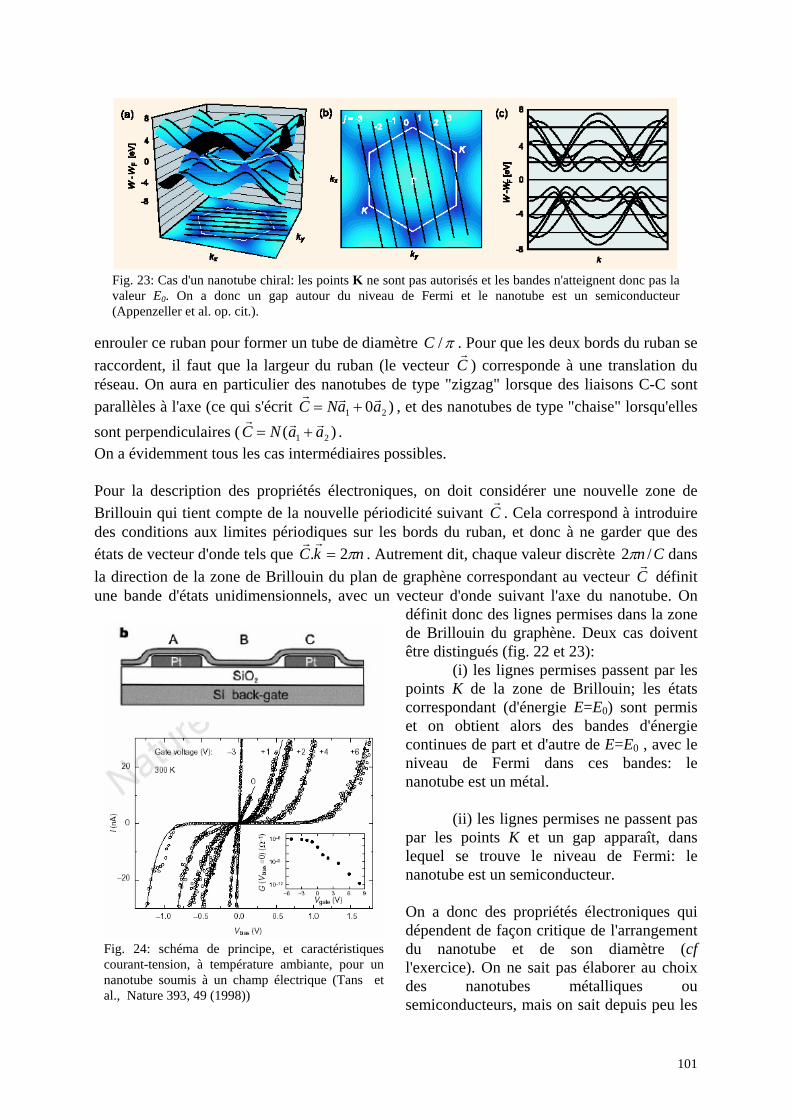
101
enrouler ce ruban pour former un tube de diamètre π/C . Pour que les deux bords du ruban se raccordent, il faut que la largeur du ruban (le vecteur C
r) corresponde à une translation du
réseau. On aura en particulier des nanotubes de type "zigzag" lorsque des liaisons C-C sont parallèles à l'axe (ce qui s'écrit )0 21 aaNC rrr
+= , et des nanotubes de type "chaise" lorsqu'elles sont perpendiculaires ( )( 21 aaNC rrr
+= . On a évidemment tous les cas intermédiaires possibles. Pour la description des propriétés électroniques, on doit considérer une nouvelle zone de Brillouin qui tient compte de la nouvelle périodicité suivant C
r. Cela correspond à introduire
des conditions aux limites périodiques sur les bords du ruban, et donc à ne garder que des états de vecteur d'onde tels que nkC π2. =
rr. Autrement dit, chaque valeur discrète Cn /2π dans
la direction de la zone de Brillouin du plan de graphène correspondant au vecteur Cr
définit une bande d'états unidimensionnels, avec un vecteur d'onde suivant l'axe du nanotube. On
définit donc des lignes permises dans la zone de Brillouin du graphène. Deux cas doivent être distingués (fig. 22 et 23): (i) les lignes permises passent par les points K de la zone de Brillouin; les états correspondant (d'énergie E=E0) sont permis et on obtient alors des bandes d'énergie continues de part et d'autre de E=E0 , avec le niveau de Fermi dans ces bandes: le nanotube est un métal. (ii) les lignes permises ne passent pas par les points K et un gap apparaît, dans lequel se trouve le niveau de Fermi: le nanotube est un semiconducteur. On a donc des propriétés électroniques qui dépendent de façon critique de l'arrangement du nanotube et de son diamètre (cf l'exercice). On ne sait pas élaborer au choix des nanotubes métalliques ou semiconducteurs, mais on sait depuis peu les
Fig. 23: Cas d'un nanotube chiral: les points K ne sont pas autorisés et les bandes n'atteignent donc pas la valeur E0. On a donc un gap autour du niveau de Fermi et le nanotube est un semiconducteur (Appenzeller et al. op. cit.).
Fig. 24: schéma de principe, et caractéristiques courant-tension, à température ambiante, pour un nanotube soumis à un champ électrique (Tans et al., Nature 393, 49 (1998))
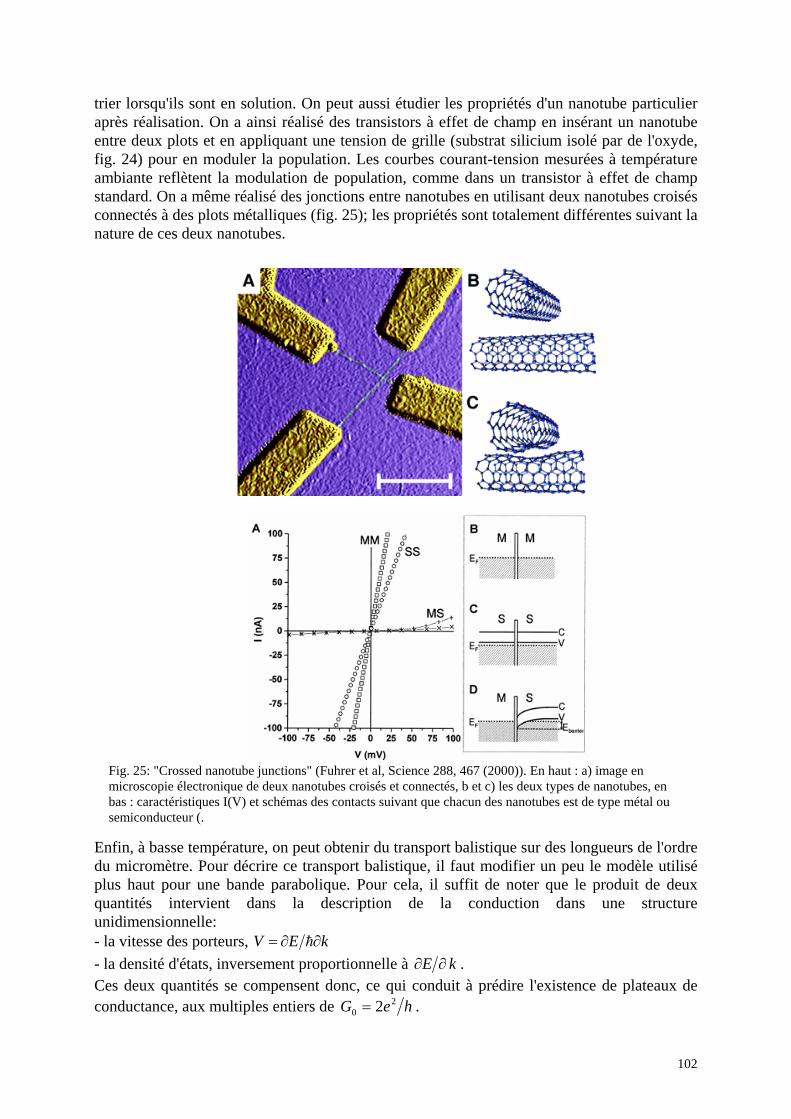
102
trier lorsqu'ils sont en solution. On peut aussi étudier les propriétés d'un nanotube particulier après réalisation. On a ainsi réalisé des transistors à effet de champ en insérant un nanotube entre deux plots et en appliquant une tension de grille (substrat silicium isolé par de l'oxyde, fig. 24) pour en moduler la population. Les courbes courant-tension mesurées à température ambiante reflètent la modulation de population, comme dans un transistor à effet de champ standard. On a même réalisé des jonctions entre nanotubes en utilisant deux nanotubes croisés connectés à des plots métalliques (fig. 25); les propriétés sont totalement différentes suivant la nature de ces deux nanotubes.
Enfin, à basse température, on peut obtenir du transport balistique sur des longueurs de l'ordre du micromètre. Pour décrire ce transport balistique, il faut modifier un peu le modèle utilisé plus haut pour une bande parabolique. Pour cela, il suffit de noter que le produit de deux quantités intervient dans la description de la conduction dans une structure unidimensionnelle: - la vitesse des porteurs, kEV ∂∂= h - la densité d'états, inversement proportionnelle à kE ∂∂ . Ces deux quantités se compensent donc, ce qui conduit à prédire l'existence de plateaux de conductance, aux multiples entiers de heG 2
0 2= .
Fig. 25: "Crossed nanotube junctions" (Fuhrer et al, Science 288, 467 (2000)). En haut : a) image en microscopie électronique de deux nanotubes croisés et connectés, b et c) les deux types de nanotubes, en bas : caractéristiques I(V) et schémas des contacts suivant que chacun des nanotubes est de type métal ou semiconducteur (.
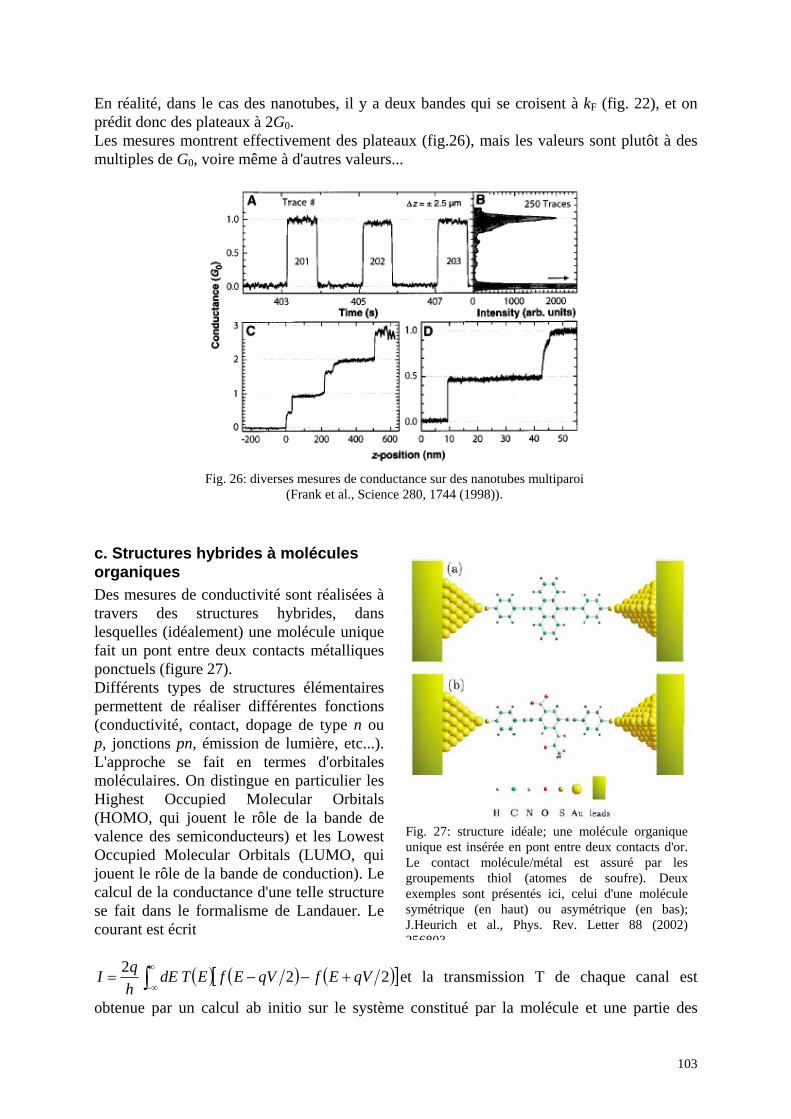
103
En réalité, dans le cas des nanotubes, il y a deux bandes qui se croisent à kF (fig. 22), et on prédit donc des plateaux à 2G0. Les mesures montrent effectivement des plateaux (fig.26), mais les valeurs sont plutôt à des multiples de G0, voire même à d'autres valeurs...
c. Structures hybrides à molécules organiques Des mesures de conductivité sont réalisées à travers des structures hybrides, dans lesquelles (idéalement) une molécule unique fait un pont entre deux contacts métalliques ponctuels (figure 27). Différents types de structures élémentaires permettent de réaliser différentes fonctions (conductivité, contact, dopage de type n ou p, jonctions pn, émission de lumière, etc...). L'approche se fait en termes d'orbitales moléculaires. On distingue en particulier les Highest Occupied Molecular Orbitals (HOMO, qui jouent le rôle de la bande de valence des semiconducteurs) et les Lowest Occupied Molecular Orbitals (LUMO, qui jouent le rôle de la bande de conduction). Le calcul de la conductance d'une telle structure se fait dans le formalisme de Landauer. Le courant est écrit
( ) ( ) ( )[ ]222 qVEfqVEfETdEhqI +−−= ∫
∞
∞−et la transmission T de chaque canal est
obtenue par un calcul ab initio sur le système constitué par la molécule et une partie des
Fig. 26: diverses mesures de conductance sur des nanotubes multiparoi
(Frank et al., Science 280, 1744 (1998)).
Fig. 27: structure idéale; une molécule organique unique est insérée en pont entre deux contacts d'or. Le contact molécule/métal est assuré par les groupements thiol (atomes de soufre). Deux exemples sont présentés ici, celui d'une molécule symétrique (en haut) ou asymétrique (en bas); J.Heurich et al., Phys. Rev. Letter 88 (2002) 256803

104
contacts métalliques. Un exemple de résultats est donné sur la fig. 28. On remarque que différents paramètres interviennent, en particulier: - la résonance avec le potentiel chimique dans les contacts; - la délocalisation des orbitales. Les orbitales π favorisent particulièrement la conduction. Pour en savoir plus, on pourra se reporter aux chapitres de Nanoelectronics and Information Technology, Rainer Waser (Ed.), Wiley.
5. Blocage de Coulomb On parle de blocage de Coulomb lorsque ce sont les interactions entre électrons qui déterminent les propriétés de transport. Les premiers résultats expérimentaux sont apparus lors de l'étude du transport électronique à travers des îlots métalliques de petite dimension: inclusions dans un matériau isolant, îlots métalliques à la surface, ou "fil quantique" interrompu. Les électrons doivent alors passer d'un îlot à l'autre, par effet tunnel. L'idée de base est qu'on ne peut faire passer un électron dans un îlot que si l'électron précédent l'a déjà quitté. Les ingrédients sont: l'effet tunnel entre les îlots, et les interactions entre électrons dans l'îlot. Le blocage de Coulomb est donc apparu d'abord plutôt comme un problème à éviter: lorsqu'il a lieu de façon non contrôlée dans un système inhomogène, il perturbe le transport électronique et introduit du bruit. Le blocage de Coulomb bien contrôlé par contre peut conduire à l'électronique à un électron. Bien sûr, c'est déjà ce que faisait Millikan en 1911 en étudiant la chute d'une goutte d'huile chargée... L'objectif est maintenant de le faire dans un système solide susceptible d'intégration en microélectronique. Le point de départ est donc un système dans lequel on pourra mettre un nombre N d'électrons en interaction. Il peut s'agir: - d'une boîte quantique de semiconducteur, telle qu'on l'a décrite au chapitre précédent, réalisée en croissance auto-organisée, par lithographie et gravure, ou délimitée dans un gaz d'électrons 2D par des contacts en surface. On sait réaliser de telles structures avec des dimensions (de la dizaine à quelques centaines de nm) du même ordre que l'inverse du vecteur d'onde de Fermi des électrons de conduction: ceux-ci occupent donc un petit nombre d'états quantifiés dans la boîte. En l'absence d'interactions, l'énergie de ces N électrons s'écrit
Φ−= ∑ =qNEU N
i iN 1, où iE est l'énergie de l'état i, et Φ le potentiel électrostatique auquel
Figure 28: résultat du calcul ab initio; J.Heurich et al., Phys. Rev. Letter 88 (2002) 256803

105
est portée la boîte. Ce potentiel Φ résulte de l'effet de champs électriques sur la boîte, et donc de la distribution des charges extérieures. On a vu à la fin du chapitre précédent que les interactions entre électrons dans la boîte étaient loin d'être négligeables et qu'un terme d'interaction doit être ajouté...mais que le calcul de ce terme n'est pas simple. - d'un îlot métallique. Pour des dimensions le plus petites qu'on sache contrôler, le nombre d'électrons libres reste grand. Autrement dit, l'inverse du vecteur d'onde de Fermi est très petit par rapport aux dimensions de l'îlot. On peut alors oublier la quantification dans l'îlot. Pour un tel système classique, l'énergie d'interaction électrostatique des N électrons est ( ) CNq 22 où C est la capacité de l'îlot, indépendante de sa charge. Il faut lui ajouter l'interaction des N électrons avec toutes les charges extérieures, représentées par le potentiel Φ. On a donc dans
ce cas classique Φ−= qNCqNU N 2
22
.
Pour discuter simplement le blocage de Coulomb, on posera Φ−+= ∑ =qN
CqNEU N
i iN 2
22
1.
Le système modèle pour l'étude du transport en régime de blocage de Coulomb comprend (fig.16) un îlot connecté à deux réservoirs (source et drain) de potentiels électrochimiques sµ et dµ . On étudie le transport à travers l'îlot en appliquant une différence de potentiel sdV , c'est-à-dire que sdsd qV=− µµ . Entre l'îlot et chacun des réservoirs, les électrons ont une probabilité non nulle de passer par effet tunnel. De plus, les électrons peuvent se réarranger de part et d'autre de la barrière tunnel (polarisation de la jonction tunnel: on crée un dipôle électrique à travers la barrière). Pour établir un circuit équivalent (fig.29), on représente ces deux mécanismes par des notions classiques, la "résistance tunnel" d'une part, la capacité de la jonction d'autre part. La résistance tunnel de la jonction est définie par le rapport entre la différence de potentiel à travers la jonction et la valeur moyenne du courant traversant la jonction. La capacité de la jonction est définie par le rapport entre la différence de potentiel à travers la jonction et la charge Q mesurant le dipôle électrique induit à travers la jonction. On contrôle le potentiel Φ de l'îlot par un contact de grille, porté à un potentiel VG, et couplé à l'îlot par une capacité CG. Cette capacité sera traitée dans le schéma électrique de la même façon que les capacités de source Cs et de drain Cd, mais les électrons ne peuvent pas passer par effet tunnel à travers le contact de grille. La tension de mesure sdV est faible et on la
µsµd
drain source
CG
R, CsR, Cd
VG
Vsd
Φ
Φ0
Fig. 29: Ilot relié à deux réservoirs d'électrons par effet tunnel, et insertion dans un circuit électrique.

106
néglige donc pour calculer l'état électrique du système à partir du schéma électrique équivalent (fig.29). Soit Φ0 le potentiel de l'îlot sans charge. Les charges des "condensateurs" sont
( )0Φ−Φ= ss CQ , ( )0Φ−Φ= dd CQ , ( )GGG VCQ −Φ−Φ= 0 , et, en faisant le bilan des courants sur l'îlot, 0=++ Gss QQQ , d'où, en posant
Gsd CCCC ++= , 0Φ+=Φ GG V
CC
.
On montre que pour un système classique, la capacité C qui intervient ici est la même que celle qui définit les interactions entre électrons dans la boîte. On a donc enfin, en posant
qCN /00 Φ= :
( )CNq
VC
CqN
CNNq
EqNCqNEU G
GN
i iN
i iN 222
20
220
2
1
22
1−−
−+=Φ−+= ∑∑ ==
Le potentiel chimique est (à température nulle) la variation de l'énergie totale du système lorsqu'on ajoute un électron. Donc
GG
NNNN VCCqNN
CqEUU −⎟
⎠⎞
⎜⎝⎛ −−+=−= − 2
10
2
1µ .
Lorsque la boîte est en contact avec les deux réservoirs à µµµ =≈ ds , le nombre d'électron se fixe à la plus grande valeur telle que µµ <N . On peut donc faire varier N en ajustant le potentiel de grille VG. On peut aussi ajouter un électron en augmentant le potentiel chimique d'une quantité ("énergie d'addition") ( ) CqEE NNNN
211 +−=−=∆ ++ µµ . Le premier terme
est la différence d'énergie entre deux niveaux: c'est ce qu'il faudrait en l'absence d'interactions. Le second est le terme classique d'interaction électrostatique. Dans cette approche simple, les deux termes s'ajoutent: dans un modèle plus élaboré, ce n'est plus le cas (voir chapitre précédent). Mais le résultat important qui reste est le suivant: pour injecter un électron dans la boîte (sur un niveau donné), il faut augmenter µ d'une énergie ∆ qui est plus grande que l'énergie nécessaire pour exciter (vers le même niveau) un électron déjà dans la boîte (fig. 30).
∆E
µ N
µ N+1
Excitation Injection
Fig. 30: Lorsque N électrons occupent l'îlot, il existe deux possibilités pour placer un électron sur le niveau excité (N+1); à gauche, on prend un électron dans la boîte et on le fait changer de niveau, ce qui coûte au moins l'énergie (EN+1-EN); à droite, on ajoute un électron dans la boîte, ce qui se fait en changeant le potentiel chimique du réservoir de la quantité (EN+1-EN+q2/C).
107
Pour une mesure de transport, on peut mesurer le courant moyen traversant l'îlot lorsqu'on applique une tension source-drain Vsd. On a deux tensions qu'on peut faire varier: la tension de grille VG et la tension source-drain Vsd. Cela conduit à deux résultats typiques: l'escalier de Coulomb, et les oscillations de Coulomb. L'analyse repose sur l'existence de l'énergie d'addition:
( ) ( ) CqEE NNNN2
11 +−=−=∆ ++ µµ Blocage de Coulomb: On mesure la conductivité en fixant Vsd à une valeur faible, ∆<sdV . La population d'électrons dans la boîte s'établit à une valeur N telle que ( ) ( )1+<≈< NN ds µµµµ . Aucun électron ne passe ni ne quitte la boîte: c'est le régime de blocage de Coulomb (fig. 31, en haut). Oscillations de Coulomb: On peut ajuster la valeur de µ par rapport à sµ et dµ grâce au potentiel de grille GV . Lorsque
( ) ( )NN sd µµµµ >>+> 1 , la première partie ( )[ ]1+> Nd µµ montre qu'un électron va
passer du réservoir ("drain") vers l'îlot, alors que la seconde partie ( ) ( )[ ]NN s µµµ >>+1 montre que l'électron va quitter aussitôt l'îlot pour rejoindre le second réservoir ("source"). On observe donc une conductivité nulle (blocage de Coulomb) sauf pour des valeurs discrètes de la tension de grille qui placent le potentiel chimique de l'îlot entre ceux des deux réservoirs (fig. 31, en bas): l'alternance de ces deux régimes lorsqu'on fait varier le potentiel de grille conduit à l'observation des oscillations de Coulomb (fig. 32). Coulomb staircase: Si on fixe la tension de grille GV , et que l'on augmente la tension de mesure sdV , on réalise une "caractéristique )(VI . En augmentant l'écart entre les potentiels chimiques des deux réservoirs, on se donne la possibilité d'y insérer un nombre croissant de
( )Nµ , et le courant augmente donc par paliers (fig. 32), l'écart entre deux paliers étant défini par E∆ . La position de ces paliers dépend de la tension de grille (ce sont les oscillations de Coulomb). Conditions d'observation
Fig. 31: Régime de blocage de Coulomb (en haut) et régime de conductance non-nulle (en bas), conduisant aux oscillations de Coulomb (Kouwenhoven et McEuen, in Trimp, Nanotechnology, Springer 1999)
Fig. 32: Oscillations de Coulomb: la conductivité est non-nulle à chaque fois que le potentiel de grille est tel que le potentiel chimique de l'îlot passe entre ceux du drain et de la source (Kouwenhoven et McEuen, in Trimp, Nanotechnology, Springer 1999
108
Pour que ces effets à un électron soient observés, deux conditions sont nécessaires: 1. L'agitation thermique ne doit pas tout brouiller: la condition est ETkB ∆< , ce qui conduit le plus souvent à travailler à basse température. De gros efforts sont faits pour réaliser des composants basés sur ces effets et fonctionnant à température ambiante... 2. Les temps caractéristiques de passage des électrons doivent être suffisamment longs. Ceci est généralement exprimé dans le langage de l'électronique: le temps caractéristique du circuit est défini à partir de la résistance tunnel et de la capacité de l'îlot: CRt=τ . La condition
Eh ∆<τ/ s'écrit CqCRh t // 2< , donc 2/ qhRt > .
Applications; le tourniquet à électron Les exemples précédents ont un inconvénient: les électrons passent un par un, mais le passage de chaque électron reste aléatoire. Des combinaisons plus ou moins compliquées associant plusieurs îlots sont imaginées pour déclencher le passage de chaque électron (transistor à un électron, pompe à électron, ...). Il en est ainsi du tourniquet à électron (fig. 21). Dans ce mode de fonctionnement, on commande le passage des électrons en modulant les barrières par deux tensions alternatives en opposition de phase: un électron passe à chaque période. On convertit ainsi un standard de fréquence en standard de courant.
Fig. 33 : Tourniquet à électron commandé par une tension alternative à 10 MHz. On pourra vérifier que les lignes pointillées correspondent à un électron par période. (Kouwenhoven et McEuen, in Trimp, Nanotechnology, Springer 1999
Fig. 32: Escalier de Coulomb dans les caractéristiques I(V). Les différentes courbes ont été enregistrées pour des tensions de grille différentes, et décalées verticalement (pour chaque courbe, I=0 si V=0). (Kouwenhoven et McEuen, in Trimp, Nanotechnology, Springer 1999

109
Exercices: 1. Contrôle de la population par un champ électrique dans une structure 2D Un exemple simple du contrôle de la population dans un gaz bidimensionnel est obtenu en insérant un puits quantique dans une structure p-i-n. Pour simplifier le calcul, on fait les hypothèses (en général raisonnables) suivantes: - dans la zone dopée p, on a un quasi-niveau de Fermi en coïncidence avec le haut de la bande de valence; - dans la zone dopée n, on a un quasi-niveau de Fermi en coïncidence avec le bas de la bande de conduction; - le champ électrique est constant dans la zone intrinsèque de part et d'autre du puits quantique; - les zones de charge d'espace (zones de désertion aux interfaces p-i et i-n, puits quantique) sont infiniment fines; - les contacts sur les zones p et n sont tels que toute la tension appliquée se retrouve dans la différence entre les quasi-niveaux de Fermi; - le courant dans la structure est négligeable; - le puits quantique est en équilibre avec la zone dopée la plus proche; - l'énergie de confinement dans le puits ne change pas. a. Repérer les conséquences de ces hypothèses sur le schéma. b. Calculer les discontinuités de champ électrique en fonction des densités surfaciques de charges en utilisant le théorème de Gauss. c. Calculer la différence entre l'énergie de l'état dans le puits et le quasi-niveau de Fermi, en fonction de la densité d'états 2D. d. Faire le bilan des charges et des variations de potentiel et en déduire la densité de porteurs dans le puits (unité: 1015m-2) en fonction de la tension appliquée. Application numérique: prendre masse effective et constante diélectrique de GaAs; le puits est à 200 nm de la zone n et à 200 nm de la zone p. Le niveau confiné est à 150 meV en dessous de la bande de conduction de la barrière. 2. Nanotubes de carbone Considérer le cas d'un nanotube "zig-zag". Montrer qu'il est soit semiconducteur, soit métal. Dans le cas semiconducteur, montrer que le gap est, en première approximation, inversement proportionnel au diamètre du nanotube. Comparer à la figure ci-contre.

110

111
Bibliographie Ne sont indiquées ici que les références un peu générales, qui vous permettront soit d'approfondir certains points de ce cours, soit au contraire d'acquérir plus de recul. Les références plus ponctuelles sont indiquées le moment venu dans chaque chapitre. Des références très générales C.Cohen-Tannoudji, B.Diu et F.Laloë, Mécanique quantique, Hermann A.Messiah, Mécanique quantique, Dunod Kittel, Introduction à la physique du solide Ashcroft et Mermin, Solid State Physics W.A.Harrisson, Electronic structures and the properties of solids, Freeman, San Francisco, 1981 Introduction: La microélectronique, où en est on? Nanoscale CMOS, Hon-Sum Philip Wong, D.J.Frank, P.M.Solomon, C.H.J.Wann, J.J.Welser, Proceedings of the IEEE 87 (1999) 537) Où va-t-on? International Technology Roadmap for Semiconductors, Semiconductor Industry Association, http://www.sematech.org/public/index.htm Première partie: Description des surfaces et croissance: Hudson, Surface Science, Wiley J.Villain et A.Pimpinelli, Physique de la croissance cristalline, Eyrolles 1995 Dislocations: J.Friedel Dislocations (Pergamon 1964) Hirth et Loth, Theory of Dislocations, (Wiley 1982). Autres sujets: D.Bimberg, M.Grundmann et N.N.Ledentsov, Quantum dot heterostructures, Wiley 1999 N.F.Nye, Physical properties of crystals: their representation by tensors ans matrices, Oxford, Clarendon Press Seconde partie: Les fonctions d'ondes dans les puits quantiques: G.Bastard, Wave mechanics applied to semiconductor heterostructures, Les Editions de Physique, Paris 1988 Revue récente sur les boîtes quantiques: D.Bimberg, M.Grundmann et N.N.Ledentsov, Quantum dot heterostructures, Wiley 1999 Blocage de Coulomb:

112
Kouwenhoven et McEuen, in Trimp, Nanotechnology, Springer 1999 Effet Hall quantique et gaz d'électrons à haute mobilité: H.Störmer, Rev. Mod. Phys 71 (1999) 875 Transport dans les nanostructures: S.Datta, Electronic transport in mesoscopic systems (Cambridge, 1999) D.K.Ferry et S.M.Goodnick, Transport in nanostructures (Cambridge University Press, 1999) Nanoelectronics and Information Technology, Rainer Waser (Ed.), Wiley

113
Février 2000 Ce problème vous propose une lecture guidée d'une partie de l'article "Mechanisms of self-ordering of quantum nanostructures grown on non-planar surfaces", par G.Biasiol et E.Kapon (Phys. Rev. Lett. 81 (1998) 2962, voir copie plus loin). Il s'agit de comprendre les principes d'une méthode d'élaboration de "fils quantiques" GaAs-Ga1-xAlxAs. Le point de départ est un substrat sur lequel on a gravé des sillons parallèles. On notera z la direction perpendiculaire au substrat, y la direction des sillons, et x la direction perpendiculaire aux deux autres. On cherche à démontrer que l'existence d'un fil quantique au fond des sillons, avec une forme stable ("self-limiting growth"), est due à deux mécanismes: - l'anisotropie de la vitesse de croissance (elle présente de fortes différences suivant l'orientation de la face exposée) - la modulation du potentiel chimique par la courbure locale de la surface et l'énergie de surface (notée γ dans l'article). On ignore deux autres mécanismes (déformation et entropie de mélange) considérés dans l'article), et on se limite à ces deux effets qui sont discutés dans le début de l'article.
L'existence d'une telle "croissance auto-régulée" permet alors d'empiler des fils de forme et taille identiques.
1. Modulation du potentiel chimique sur une surface gravée
Fig.2: section (microscopie électronique en transmission) de deux échantillons présentant un empilement de "fils quantiques". Le GaAs apparaît en sombre, le GaAlAs en plus clair. Les fils quantiques apparaissent dans l'angle inférieur des "V" de GaAs; ils sont repérés "QWRs" pour "quantum wires".
z
x
Fig.1: image (section, en microscopie électronique en transmission) d'un fil quantique unique (QWR=quantum wire). Ce fil a été dopé de façon sélective: des donneurs dans les barrières (notés +) permettent de réaliser dans le fil quantique un gaz d'électrons unidimensionnel (noté - ).

114
a. On considère un substrat dont la surface présente une modulation ),( yxz . On suppose dans un premier temps que l'énergie de surface dépend de l'orientation de façon régulière (hypothèse haute température), et on cherche à établir une partie de l'équation (1) de l'article. Montrer que la formule de Herring-Mullins permet d'écrire le potentiel chimique local sous la forme (c'est une partie de l'équation 1 de l'article) [ ]yx κκγµ +Ω+ ~
00 où 0µ est le potentiel
sur la surface plane de référence, 0Ω est le volume élémentaire dans le solide, et 2
2
xz
x ∂∂
−=κ
représente la courbure locale dans la direction x (respectivement y). Application: substrat sur lequel on a gravé un réseau sinusoïdal, )/2sin(),( λπ yAyxz = . Soit ρ la densité d'adatomes. Calculer le courant d'adatomes dû à la modulation du potentiel.
On rappelle la relation de Nernst-Einstein µρ ∇−=rr
TkD
jB
s .
Montrer que ce courant d'adatomes tend à diminuer la courbure locale. b. La modélisation proposée ensuite dans l'article suppose que la surface gravée présente des facettes correspondant aux singularités de l'énergie de surface. L'expression ci-dessus du potentiel chimique doit donc être écrite pour le cas d'une surface facettée: c'est l'équation 2 de l'article, qu'on va ré-établir maintenant. Rappeler la construction de Wulff dans le cas de facettes, et écrire le potentiel chimique à la surface d'une cristallite convexe, ayant sa forme d'équilibre, en fonction des distances ih au point de Wulff. Ré-écrire ce potentiel chimique, pour une facette rectangulaire de côté xl (et
yl ), limitée par des facettes faisant avec elle un angle xθ (et yθ ) aigu, sous la forme
⎥⎥⎦
⎤
⎢⎢⎣
⎡+Ω+
y
y
x
x
llγγ
µˆˆ
00 où les numérateurs sont des fonctions des énergies de surface et des
angles entre les facettes qu'on explicitera. Que se passe-t-il si la surface est concave? En déduire la variation du potentiel chimique pour la surface gravée de sillons facettés ci-dessous, dans le cas tbs lll ,>> , puis dans le cas général. Comparer à l'expression donnée dans l'article (eq. 2).
2. Evolution de la surface en l'absence de diffusion. Dans la méthode d'élaboration utilisée (MOCVD, Metal-Organic Chemical Vapor Deposition), les vitesses de croissance résultent de réactions chimiques locales entre la surface du substrat et la phase vapeur. On a alors des vitesses de croissance très différentes suivant l'orientation de la face de croissance. Soient R la vitesse de croissance mesurée sur
Fig.3: schéma des sillons facettés;et potentiel chimique local; le fond des sillons a une largeur lb ("bottom"), les crêtes une largeur lt ("top") et les côtés une largeur ls ("sidewall"). On note x la direction horizontale, z la direction verticale, y la direction perpendiculaire à la feuille (axe des sillons). Les fonds et les crêtes sont des facettes orientées (100), les côtés sont orientés (111). On néglige une petite facette (311) signalée sur la fig.2

115
une face (100) plane, et RrR ss = , RrR bb = , RrR tt = les vitesses de croissance suivant z sur des facettes de grande taille. On note τ le temps moyen passé par un atome sous forme d'adatome (avant son incorporation: c'est donc le temps pendant lequel il reste mobile à la surface). On suppose que τ est indépendant de l'orientation. On suppose que les adatomes ne diffusent pas (à l'échelle de la taille des sillons). Exprimer les vitesses de croissance dans la direction z sur les différentes facettes. Exprimer la densité d'adatomes (projetée dans le plan x,y) sur les différentes facettes. Des mesures préliminaires on montré que tbs rrr ,> . Calculer la vitesse d'expansion ou de disparition des deux types de facettes (100), sommet et fond des sillons. 3. Effet de la diffusion. On suppose maintenant une forte diffusion, c'est-à-dire que la longueur de diffusion des adatomes τsdif Dl = est grande devant bt ll , . Le calcul du courant d'adatomes demande une résolution détaillée des équations d'évolution. On va se contenter d'une approximation utilisant la relation de Nernst-Einstein, où on remplace le gradient par ( ) tbtbs l ,, /µµ − (remarque: à la limite de deux facettes semi-infinies, ce serait ( ) diftbs l/,µµ − ). Etablir l'expression (3) de l'article (on identifiera et on justifiera les approximations nécessaires). Montrer qu'on peut obtenir des facettes de largeur stable ("auto-régulée") en fond de sillon: on dira qu'une facette est stable si sa largeur reste constante pendant la croissance. Montrer qu'une longueur de diffusion plus grande sur GaAs que sur GaAlAs permet d'envisager de réaliser un "fil quantique". 4. Confinement. On réalise des fils quantiques par la méthode précédente. Commenter la forme observée (Fig. 1 ci-dessus) par rapport à la description précédente: en particulier, forme du fond de sillon, étranglement au bord du fil. Pour modéliser le confinement, on "déplie" le fond du sillon (et le "V" du GaAs). Evaluer sur la figure les dimensions caractéristiques du fil ainsi obtenu. Calculer les énergies de confinement dans les deux directions orthogonales, dans l'approximation de barrières infiniment hautes. Dans quelle mesure l'approximation de barrières infiniment haute est-elle justifiée (on discutera le confinement suivant la hauteur du fil (axe z) et suivant sa largeur)? Tracer la densité d'états (en cm-1 meV-1) sur les premiers niveaux. La masse effective dans GaAs est 0
* 067.0 mm = . Le décalage de bande de conduction entre GaAs et Ga0.7Al0.3As est meV 2500 =V . On rappelle aussi les constantes usuelles
kg 109
JK 1038.1C 106.1
Js 10054.1
310
1-23
19
34
−
−
−
−
×=
×=
×=
×=
mke
B
h

116
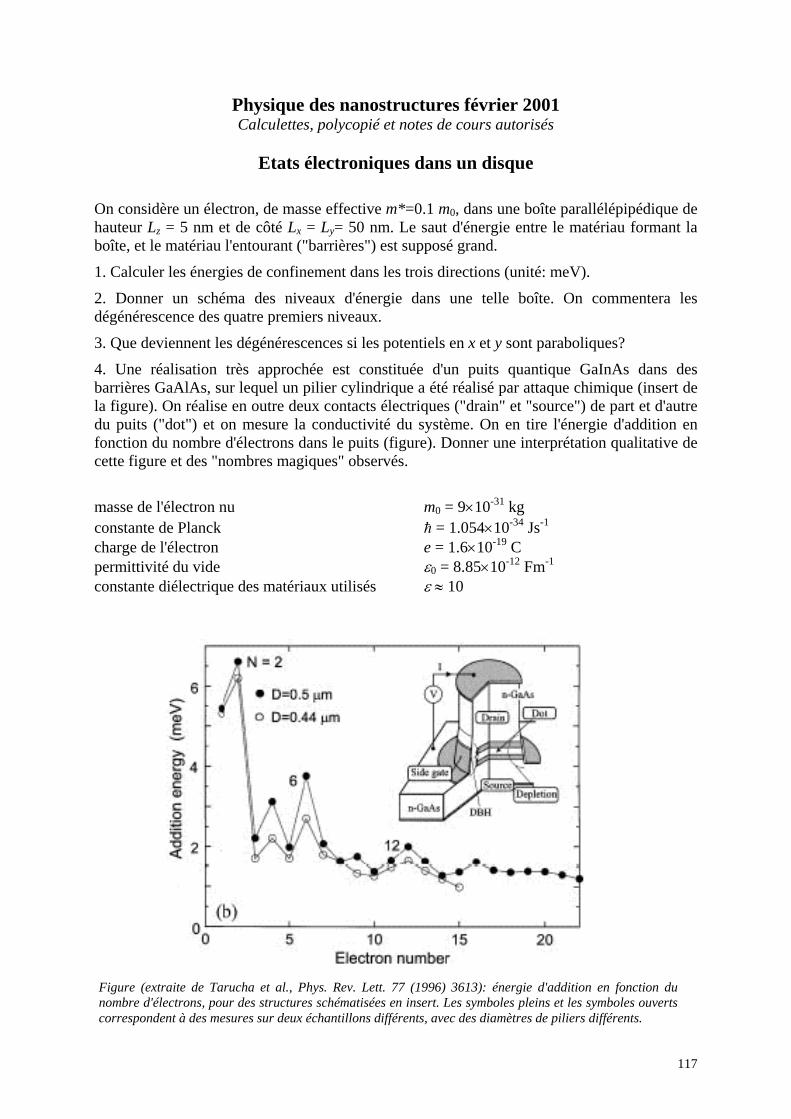
117
Physique des nanostructures février 2001 Calculettes, polycopié et notes de cours autorisés
Etats électroniques dans un disque
On considère un électron, de masse effective m*=0.1 m0, dans une boîte parallélépipédique de hauteur Lz = 5 nm et de côté Lx = Ly= 50 nm. Le saut d'énergie entre le matériau formant la boîte, et le matériau l'entourant ("barrières") est supposé grand.
1. Calculer les énergies de confinement dans les trois directions (unité: meV).
2. Donner un schéma des niveaux d'énergie dans une telle boîte. On commentera les dégénérescence des quatre premiers niveaux.
3. Que deviennent les dégénérescences si les potentiels en x et y sont paraboliques?
4. Une réalisation très approchée est constituée d'un puits quantique GaInAs dans des barrières GaAlAs, sur lequel un pilier cylindrique a été réalisé par attaque chimique (insert de la figure). On réalise en outre deux contacts électriques ("drain" et "source") de part et d'autre du puits ("dot") et on mesure la conductivité du système. On en tire l'énergie d'addition en fonction du nombre d'électrons dans le puits (figure). Donner une interprétation qualitative de cette figure et des "nombres magiques" observés.
masse de l'électron nu m0 = 9×10-31 kg constante de Planck h = 1.054×10-34 Js-1 charge de l'électron e = 1.6×10-19 C permittivité du vide ε0 = 8.85×10-12 Fm-1 constante diélectrique des matériaux utilisés ε ≈ 10
Figure (extraite de Tarucha et al., Phys. Rev. Lett. 77 (1996) 3613): énergie d'addition en fonction du nombre d'électrons, pour des structures schématisées en insert. Les symboles pleins et les symboles ouverts correspondent à des mesures sur deux échantillons différents, avec des diamètres de piliers différents.

118
Evolution d'un îlot pyramidal à la surface d'un cristal
Ce sujet est inspiré de l'article intitulé "A novel method to determine the Ehrlich-Schwöbel barrier", présenté par R.Gerlach, T.Maroutian, L.Douillard, D.Martinotti et H.J.Ernst au "2nd LEEM/PEEM workshop, Paris (2000), et à paraître dans la revue Surface Science. Les figures 1 et 4 ci-dessous sont extraites de cet article.
Les données sont obtenues par l'observation en LEEM (Low Energy Electron Microscopy) de la surface d'un cristal de Cu d'orientation (001). Cette méthode de microscopie, basée sur la diffraction d'électrons de faible énergie, permet d'obtenir des images de la surface d'un cristal, et en particulier de son organisation en terrasses et en marches. Une préparation de la surface a permis de former des îlots pyramidaux, quasi-circulaires, épais de plusieurs monocouches, mais dont le sommet est formé d'une terrasse plate d'orientation (001). Les flancs de tels îlots sont formés de marches très serrées. Pour permettre des calculs simple, on supposera que cet îlot épais initial ("pyramide") est parfaitement circulaire, de rayon R, et que ses flancs sont quasi-verticaux.
On a enregistré l'évolution de la pyramide en présence d'un flux incident F d'atomes de Cu. Les atomes de Cu incidents sont adsorbés sur la terrasse formant le sommet de la pyramide, où ils diffusent. Dans les cas favorables, ils créent un centre de nucléation au voisinage du centre de la terrasse sommitale; ce noyau critique évolue ensuite en un îlot d'épaisseur égale à
une monocouche, plus ou moins concentrique à la terrasse sommitale, et dont le rayon ρ augmente. Lorsque ce rayon ρ est assez grand, un autre noyau critique se forme au voisinage du centre et donne naissance à un deuxième îlot, qui croît, et ainsi de suite. On observe alors un train de marches plus ou moins concentriques qui se déplacent, au cours du temps, vers le bord de la pyramide. La fig.1a est une observation à un instant donné, en LEED, où les marches apparaissent sous forme de lignes plus sombres.
Dans l'approximation utilisée (fig. 1b), les marches sont parfaitement circulaires. Ces marches sont numérotées à partir du centre de la terrasse, leurs rayons sont
notés ρ1, ρ2, ... ρn.
La figure 1c représente la densité d'adatomes sur les terrasses entre deux marches, calculée dans le cadre du modèle Burton-Cabrera-Franck. On néglige toute influence de la structure cristalline dans les deux directions du plan (001). Dans ce qui suit, on va faire ce calcul et en déduire la vitesse de déplacement des marches et donc, dans des cas simples, l'évolution dans le temps de ρn(t) pour les différentes marches. On utilisera ce calcul pour interpréter les mesures réalisées.

119
I. Cas sans barrière Schwöbel:
On suppose, dans cette première partie, que tout adatome arrivant à une marche, que ce soit par en haut ou par en bas, y est incorporé.
1. On note a la distance inter-atomique en surface (c'est-à-dire, le pas de la maille carrée), ν la probabilité (par unité de temps) de saut d'un site au site proche voisin, D le coefficient de diffusion, ( )tr ,rη la densité d'adatomes au point rr à l'instant t, F le flux incident; écrire l'équation d'évolution de η sur une terrasse en négligeant la désorption et en supposant que l'incorporation se fait seulement en bord de marche.
2. On note ηequ la densité des adatomes à l'équilibre. Discuter les propriétés de symétrie et les conditions aux limites imposées à la densité d'adatomes en régime permanent ( )rrη : - pour l'îlot central de rayon ρ 1; - pour la terrasse circulaire située entre deux marches de rayons ρ (n-1) et ρ n.
3. On rappelle l'expression du Laplacien à deux dimensions en coordonnées polaires (r,θ)
2
2
22
2
2
2
2
2 11θ∂∂
+∂∂
+∂∂
=∂∂
+∂∂
=∆rrrryx
.
Calculer la densité d'adatomes ( )rrη sur l'îlot central en régime permanent.
4. Calculer la densité d'adatomes sur une terrasse circulaire entre deux marches. Commenter les différences par rapport à la fig1c.
5. Etablir l'expression de la vitesse de déplacement d'une marche, en fonction des rayons des différentes marches (On exprimera les vitesses à partir des courants d'adatomes au voisinage de la marche, que l'on explicitera ensuite à partir du résultat de la question précédente: on devra distinguer le cas de la marche centrale et celui des autres marches).
6. On se place dans le cas où un seul îlot s'est formé sur la terrasse sommitale. On note simplement ρ le rayon de l'unique marche. Déduire de la question précédente une équation différentielle pour l'évolution de ρ(t).
7. Montrer que la résolution de cette équation différentielle permet d'écrire le temps t de dépôt passé entre la nucléation de l'îlot et son extension à un rayon ρ, en utilisant la fonction
( ) duuuf
X
∫ −−=
2
0 1Ln .
Montrer que ( )∑∞
=
−−=
1 2
211)(n
n
nXXf .
La fonction )(Xf est tracée fig.2. En déduire un tracé de ρ(t).
8. Calculer directement (à partir de la question 6) la vitesse de la marche en ρ =0 et ρ = R.
9. Que pensez-vous du modèle au voisinage de ρ =0?
0.0 0.2 0.4 0.6 0.8 1.00
1
f(X)
X
Fig2: tracé de la fonction de la question 7.
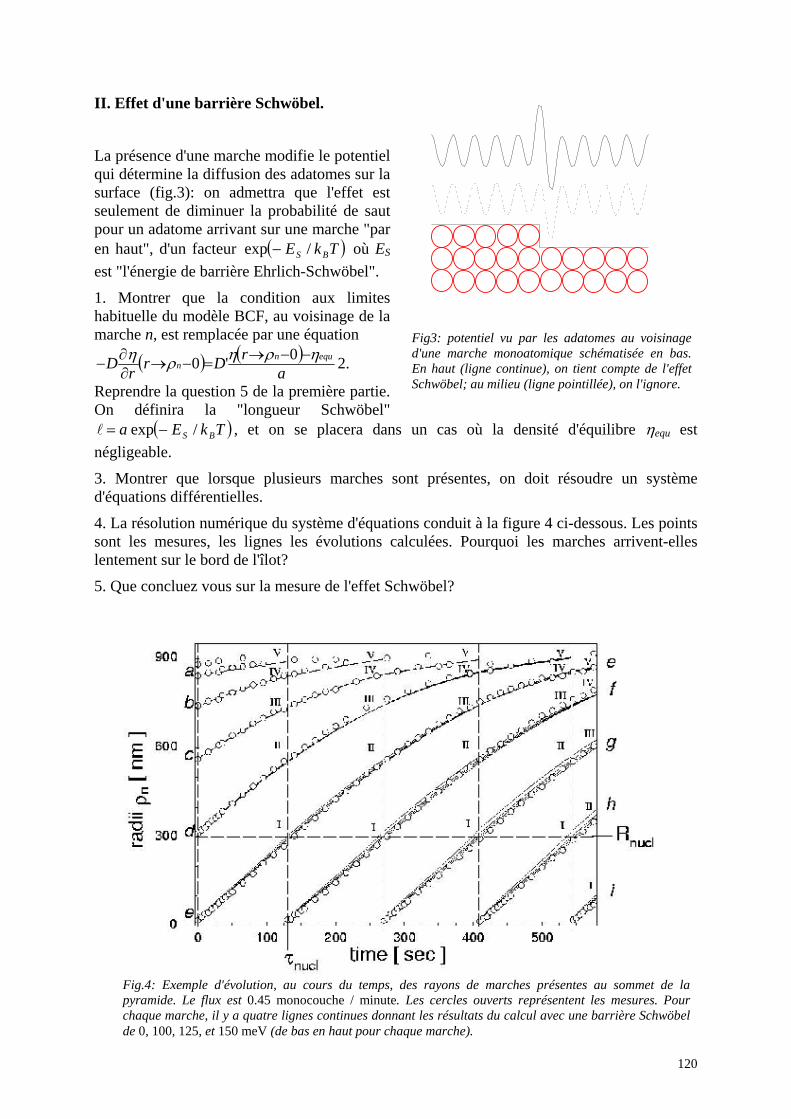
120
II. Effet d'une barrière Schwöbel.
La présence d'une marche modifie le potentiel qui détermine la diffusion des adatomes sur la surface (fig.3): on admettra que l'effet est seulement de diminuer la probabilité de saut pour un adatome arrivant sur une marche "par en haut", d'un facteur ( )TkE BS /exp − où ES est "l'énergie de barrière Ehrlich-Schwöbel".
1. Montrer que la condition aux limites habituelle du modèle BCF, au voisinage de la marche n, est remplacée par une équation
( ) ( )a
rDrrD equnn
ηρηρη −−→=−→∂∂− 0'0 2.
Reprendre la question 5 de la première partie. On définira la "longueur Schwöbel"
( )TkEa BS /exp −=l , et on se placera dans un cas où la densité d'équilibre ηequ est négligeable.
3. Montrer que lorsque plusieurs marches sont présentes, on doit résoudre un système d'équations différentielles.
4. La résolution numérique du système d'équations conduit à la figure 4 ci-dessous. Les points sont les mesures, les lignes les évolutions calculées. Pourquoi les marches arrivent-elles lentement sur le bord de l'îlot?
5. Que concluez vous sur la mesure de l'effet Schwöbel?
Fig3: potentiel vu par les adatomes au voisinage d'une marche monoatomique schématisée en bas. En haut (ligne continue), on tient compte de l'effet Schwöbel; au milieu (ligne pointillée), on l'ignore.
Fig.4: Exemple d'évolution, au cours du temps, des rayons de marches présentes au sommet de la pyramide. Le flux est 0.45 monocouche / minute. Les cercles ouverts représentent les mesures. Pour chaque marche, il y a quatre lignes continues donnant les résultats du calcul avec une barrière Schwöbel de 0, 100, 125, et 150 meV (de bas en haut pour chaque marche).

121
Février 2002 Premier problème: corral quantique.
La figure ci-contre représente un "corral quantique": des d'atomes de fer (Fe) ont été déposés sur la surface (111) d'un cristal de cuivre (Cu), et mis en place à l'aide d'une pointe STM (scanning tunneling microscope) pour former un cercle. La surface est ensuite étudiée à l'aide de la même pointe STM. Les atomes apparaissent comme un cercle de plots. Les ondes stationnaires à l'intérieur du "corral" sont attribuées aux états électroniques à la surface du cuivre. Ce sont ces états que l'on va décrire dans la suite.
On note z la normale à la surface, x et y deux directions du plan. L'hypothèse de travail est la suivante: (i) confinement dans la direction z: à la surface du cuivre, les électrons sont confinés dans l'état fondamental d'un puits de potentiel de très faible épaisseur (typiquement sur une distance inter-atomique à la surface du cuivre); les hauteurs de barrières sont très grandes; le mouvement libre dans le plan x,y est alors décrit par une masse effective m* = 0.38 m0; (ii) confinement dans le plan x,y: les atomes de fer créent un potentiel de confinement latéral (directions x et y), en forme de disque de rayon R, également avec des barrières très hautes. 1. Justifier que l'on cherche les fonctions enveloppes sous la forme ( ) ( )θ,rFzf . 2. Evaluer les ordres de grandeur des énergies dues au confinement dans la direction z et dans les directions x et y du plan si R = 7.1 nm (exprimer ces énergies en meV). 3. Ecrire l'hamiltonien et montrer qu'il commute avec l'opérateur projection du moment cinétique Lz dans la direction z. 4. En déduire que la fonction F peut être cherchée sous la forme ( ) ( ) ( )θθ imrRrF exp, = et préciser les valeurs possibles de m. 5. Ecrire l'équation différentielle que doit vérifier la fonction R(r). 6. Au vu de cette équation, que pouvez-vous dire de la dégénérescence (spin non compris) des niveaux d'énergie? 7. Montrer que les énergies propres s'expriment à partir des zéros des fonctions de Bessel (définies ci-dessous). 8. Calculer (en meV), les énergies des premiers niveaux obtenus en utilisant les zéros de la fonction de Bessel J0. 9. On peut utiliser la pointe STM dans un mode spectroscopique en enregistrant le signal en fonction de la tension V appliquée entre la pointe et l'échantillon. Pour un corral de rayon

122
R = 7.1 nm, en plaçant la pointe au centre du corral, on a mesuré des pics aux valeurs suivantes de V:
-425 mV, -370 mV, -274 mV, -148 mV, 0, +170 mV.
(M.F.Crommie, C.P.Lutz, D.M.Eigler, Science 262, 218 (1993)) Interpréter ces mesures à partir des résultats de la question précédente (pour aborder la question, on pourra tracer la suite des tensions mesurées en fonction de la suite des énergies calculées en 8). On néglige l'effet éventuel des interactions entre électrons. 10. Lorsqu'on place la pointe à côté du centre du corral, on voit apparaître d'autres pics. A quoi les attribueriez-vous?
• • • • • • • • • • • • • • •
On rappelle les constantes usuelles
kg 109C 106.1
Js 10054.1
310
19
34
−
−
−
×=
×=
×=
meh
On rappelle l'expression du Laplacien en coordonnées cylindriques:
2
2
2
2
22
2
2
2
2
2
2
2 11zrrrrzyx ∂∂
+∂∂
+∂∂
+∂∂
=∂∂
+∂∂
+∂∂
θ.
Fonctions de Bessel: les solutions de l'équation différentielle
0²²1''' =⎥⎦⎤
⎢⎣⎡ −++ y
xm
xyy
sont les combinaisons linéaires des fonctions de Bessel Jm et Ym. La fonction Jm est la seule solution bornée, alors que la fonction Ym diverge en x = 0. La fonction Jm est donnée ci-dessous pour les premières valeurs entières de m. On notera que J0 est la seule qui prenne une valeur non-nulle à l'origine.
Les zéros des premières fonctions de Bessel (valeurs de x telles que Jm(x) = 0) sont: - pour J0 : x = 2.405 5.520 8.654 11.79 14.93 18.07 etc... - pour J1 : x = 3.832 7.016 10.173 13.32 16.47 etc...
• • • • • • • • • • • • • • •

123
Second problème: croissance sur une surface vicinale: nucléation sur les terrasses ou croissance par avancée de marches? On souhaite estimer la probabilité de nucléation d'îlots sur les terrasses, par rapport à la croissance par avancée de marches, sur un substrat d'orientation légèrement désorientée par rapport à une surface de haute symétrie. L'angle de désorientation est 2°. On utilise dans la suite d'une description simplifiée de la surface qui pourrait s'appliquer à un substrat GaAs. On note z la direction normale à la surface, et on suppose que la surface est bien décrite par un réseau de marches rectilignes, suivant la direction y, de hauteur h = 0.28 nm, régulièrement réparties dans la direction perpendiculaire notée x. Le substrat est de type "solid-on-solid" adapté (réseau carré de côté a = 0.4 nm dans les directions x et y, hauteur h = 0.28 nm dans la direction z). La croissance correspond à un seul type d'atomes ou molécules, de flux incident F, et on se place dans des conditions de croissance telles que tous les atomes (ou molécules) incidents s'adsorbent, et que toute ré-évaporation des adatomes est négligeable. Le flux F correspond à une croissance d'une monocouche par seconde. I. Croissance par avancée de marche 1. Calculer la largeur L des terrasses et la vitesse d'avancée des marches. 2. Dans le modèle BCF, écrire l'équation différentielle que suit la densité d'adatomes ρ en utilisant les hypothèses simplificatrices ci-dessus. 3. Résoudre cette équation différentielle simplifiée, et en supposant une incorporation totale aux bords de marche avec une densité d'adatomes en bord de marche ρ0. 4. La constante de diffusion en surface des adatomes est donnée par l'expression
( )TkEaD D B2 exp
21
−= ν ,
où ν ≈ 1013 s-1 est une fréquence caractéristique de vibration des atomes et ED ≈ 1 eV est l'énergie d'activation (barrière d'énergie à franchir pour sauter d'un site au site voisin). Le substrat est maintenu à la température 550 °C. Tracer la variation de la densité d'adatomes sur les terrasses et calculer la densité maximale (en adatomes par cm²). On supposera que ρ0 est négligeable. II. Nucléation homogène. L'approche la plus simple de la nucléation d'îlots consiste à considérer une distribution homogène d'îlots sur la surface (densité ni d'îlots de taille i). On suppose que seuls les adatomes (i = 1) sont mobiles, et qu'ils s'agrègent de façon irréversible avec les autres adatomes et sur les îlots plus gros. 1. Justifier les équations d'évolution suivantes:

124
iiiii
iii
nDnnDndt
dn
dtdn
nDnnDFdt
dn
σσ
σσ
1111
2
21
211
1
...
...
2
−=
=
−−=
−−
≥∑
2. Les "coefficients de capture" iσ doivent être calculés en tenant compte de la taille et de la forme des îlots, et de leur environnement (îlots voisins...). Dans une approche très simplifiée, on pose iσ = 1 pour tout i. Ecrire les équations d'évolution de n1 et ∑
≥
=2i
inN .
3. On reprend le cas de la croissance sur substrat vicinal. On suppose que la croissance se fait principalement par avancée de marche, la densité d'adatomes étant décrite par le modèle BCF. A partir de cette densité d'adatomes, et pour les conditions de croissance de la première partie, évaluer la probabilité de nucléation d'îlots sur les terrasses en cours de croissance, en utilisant l'hypothèse de la nucléation homogène simplifiée. Quel critère proposeriez-vous pour estimer l'importance de ce mécanisme? Conclure.
• • • • • • • • • • • • • • •
On rappelle les constantes usuelles
kg 109
JK 1038.1C 106.1
Js 10054.1
310
1-23
19
34
−
−
−
−
×=
×=
×=
×=
m
ke
B
h

125
Morphologie de la croissance en épitaxie par jets moléculaires: paramètres clés pour la formation de boîtes quantiques
(tiré de l'article de F.Tinjod et al, "Key parameters for the formation of self-assembled quantum dots", compte-rendu de la conférence 2002 de la European Materials Research Society) Cet article discute les paramètres gouvernant le mode de relaxation de la déformation élastique lors de l'épitaxie d'un matériau semi-conducteur présentant un désaccord de paramètre de maille par rapport au substrat. Ils calculent l'énergie de la couche, à l'équilibre, pour différents types de morphologie. Ils appliquent les résultats obtenus à l'épitaxie de InAs sur GaAs, CdTe sur ZnTe, CdSe sur ZnSe, le substrat étant orienté (001). On note:
h l'épaisseur moyenne de la couche déposée (faible par rapport à l'épaisseur du substrat), ( ) ccs aaa −=maxε la déformation dans le plan de la couche en épitaxie cohérente,
d0 la distance moyenne entre dislocations pour une relaxation complète de la couche, γ l'énergie surfacique, par unité de surface (001), de la couche déposée.
1. On a montré en cours que l'énergie de la couche en épitaxie cohérente d'épaisseur uniforme h, écrite par unité de surface, se met sous la forme ( ) γε += hMhE 2
maxcoh-2D . Discuter cette expression en cinq lignes au plus. 2. On suppose d'abord que la relaxation a lieu par introduction à l'interface d'un réseau carré de dislocations. Pour des couches de structure zinc-blende orientées (001), la projection sur l'interface du vecteur de Burgers des dislocations les plus fréquemment observées est
22/ab = . L'énergie par unité de longueur d'une dislocation est notée ED. Ecrire la distance moyenne d0 entre dislocations pour une couche complètement relaxée, et l'énergie du double réseau de dislications, par unité de surface de la couche complètement relaxée, qu'on notera E0. La calculer (en meV Å-2) pour les deux couples de matériaux du tableau ci-dessous. 3. Définir une épaisseur caractéristique h0 permettant de comparer l'énergie de la couche cohérente et celle de la couche complètement relaxée, et la calculer pour les deux couples de matériaux envisagés. 4. On envisage maintenant le cas d'une couche partiellement relaxée par un réseau de dislocations avec une distance moyenne d. Exprimer l'énergie de la couche (par unité de surface) dans cette configuration. On utilisera comme paramètres h0, d0, E0 et γ . On notera cette énergie E2D,disl (h,d). 5. Montrer que, pour une épaisseur h donnée, il existe une valeur de d(h) pour laquelle E2D,disl (h,d) est minimale. Donner la condition pour que ce minimum ait une signification physique. Ecrire alors l'énergie minimum, qu'on notera E2D,disl (h). 6. Tracer les trois énergies E2D,coh (h), E2D,disl (h), et l'énergie de la couche complètement relaxée, et en déduire qu'il existe une épaisseur critique hc qui correspond au passage d'une couche 2D cohérente à une couche 2D avec dislocations. 7. La calculer pour les deux matériaux.

126
8. On envisage maintenant la formation d'îlots épais sur une couche de mouillage (croissance Stranski-Krastanov). On suppose que la forme et la taille des îlots obtenus sont fixées par les propriétés du matériau et les conditions de croissance. Justifier en quelques lignes l'expression de l'énergie
( ) ( ) ( )βγεα ++−= 11 2maxcoSK, hMhE h .
On ne calculera pas α et β mais on justifiera les signes choisis. 9. Des calculs numériques permettent de calculer α pour des îlots de forme pyramidale: on trouve qu'une valeur α = 0.4 est une bonne approximation. Tracer E2D,coh (h) et ESK,coh (h), et en déduire qu'il existe une épaisseur critique hSK qui correspond au passage d'une couche 2D cohérente à une couche avec des îlots mais sans dislocations. 10. La calculer pour les deux couples de matériaux. 11. Conclure sur le mode de relaxation attendu pour chaque couple de matériaux, et discuter le résultat à partir des trois énergies mises en jeu (élastique, dislocation, surface). 12. On considère un puits quantique CdTe inséré dans du ZnTe. La bande interdite de CdTe est 1.606 eV, celui de ZnTe est 2.391 eV. Cet écart est réparti (figure 1) sous la forme d'un décalage de la bande de valence (40 % de la différence de bande interdite) et de la bande de conduction (60 %) en sens opposés. Les masses effectives sont 0.65 m0 pour les trous au voisinage du haut de la bande de valence et 0.11 m0 pour les électrons au voisinage du bas de la bande de conduction. Calculer l'énergie de confinement ∞
1E pour un électron de conduction dans un puits CdTe de largeur 17 Å, ainsi que pour un trou. 13. Utiliser la figure 2 pour en déduire les énergies de confinement de l'électron ( eE1 ) et du trou ( hE1 ) dans un puits formé par une couche de 17 Å de CdTe dans du ZnTe. En déduire l'énergie de transition repérée sur la figure 1 par la flèche "PL". On retranchera 20 meV à la valeur calculée pour tenir compte de l’interaction électron-trou (formation d’un exciton). 14. Un échantillon a été élaboré en insérant plusieurs puits CdTe d'épaisseur croissante dans du ZnTe, et on a mesuré le spectre de luminescence de la figure 3. Identifier les raies observées, tracer l'énergie de transition mesurée en fonction de l'épaisseur du puits quantique; pointer sur la même figure le résultat de la question 13. Commenter: on pourra discuter l'intensité des raies et leur position. 15. Imaginez un échantillon du même type, à base de GaAs et InAs. Le matériau de petite bande interdite est InAs. Quelles épaisseurs choisissez-vous et qu'attendez-vous comme spectre (intensités et positions des raies)? matériaux a (Å) M (GPa) ED (eV/ Å) βγ (meV/ Å2) InAs / GaAs 6.058 / 5.653 80 1 5 CdTe / ZnTe 6.481 / 6.104 40 0.55 10
127
On rappelle les constantes usuelles
kg 109
JK 1038.1C 106.1
Js 10054.1
310
1-23
19
34
−
−
−
−
×=
×=
×=
×=
m
ke
B
h
EG(ZnTe) EG(CdTe)
E1e
E1h
PL
ZnTe CdTe ZnTe Figure 1
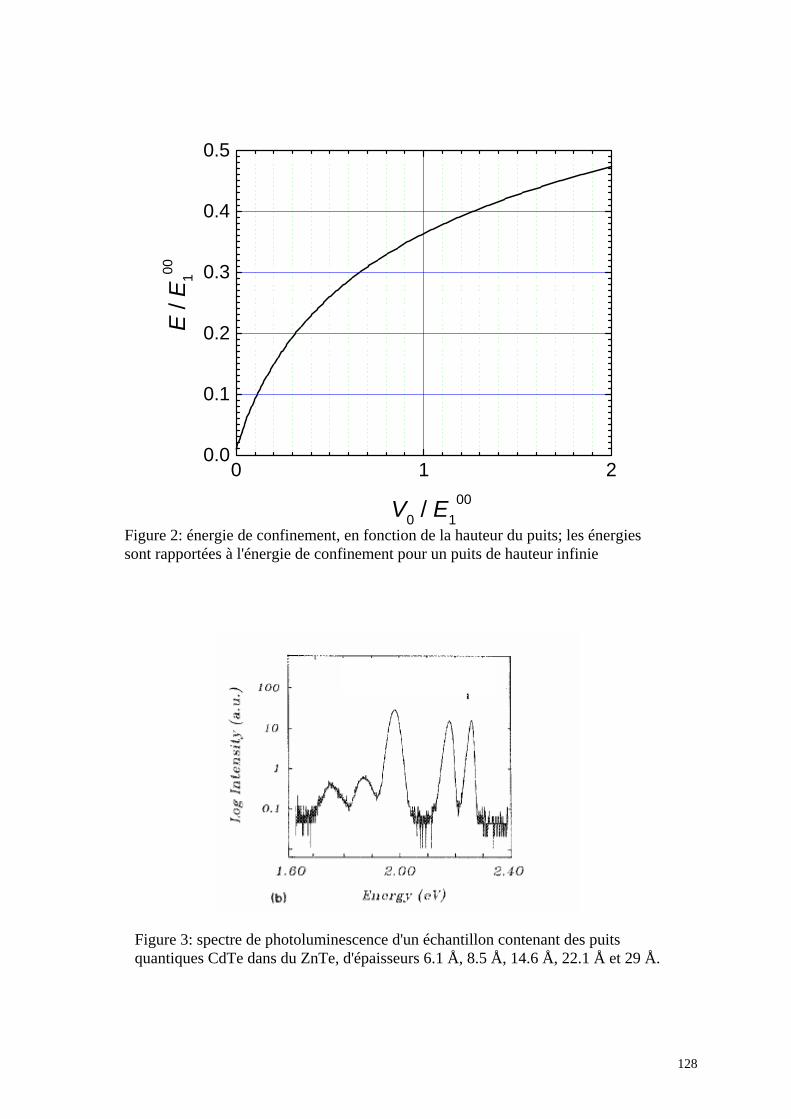
128
0.0
0.1
0.2
0.3
0.4
0.5
0 1 2
V0 / E100
E / E
100
Figure 2: énergie de confinement, en fonction de la hauteur du puits; les énergies sont rapportées à l'énergie de confinement pour un puits de hauteur infinie
Figure 3: spectre de photoluminescence d'un échantillon contenant des puits quantiques CdTe dans du ZnTe, d'épaisseurs 6.1 Å, 8.5 Å, 14.6 Å, 22.1 Å et 29 Å.
129
I. Energie d'échange en confinement à 0D (tiré de G.Lamouche et G.Fishman, "Two interacting electrons in a three-dimensional parabolic quantum dot: a simple solution", J. Phys.: Condens. Matter 10 (1998) 7857. On considère des îlots sphériques de semiconducteur, avec comme masse effective m* = 0.11 m0 et comme constante diélectrique ε = 9.8 (paramètres de CdTe).
L'utilisation d'un potentiel de confinement parabolique permet un calcul simple de l'interaction de deux électrons confinés dans la même boîte. On utilise le potentiel représenté ci-contre; l'origine des énergies correspond à l'énergie minimale dans la barrière, et le potentiel dans la boîte pour un électron de masse effective m est donné par
( ) 22*21
0 rmVrV ω+−=r .
On prendra V0 = 1 eV.
Les états propres de l'oscillateur harmonique à une dimension sont rappelés ci-dessous. On pose ω*0 mhl = . 1. Calculer la valeur de 0l correspondant à un écart 1s-1p ∆ = 100 meV. 2. On place deux électrons dans la boîte. Ecrire l'équation différentielle décrivant le mouvement relatif des deux électrons, en tenant compte du potentiel de confinement et de l'interaction électrostatique.
3. Dans un premier temps, on ne tient pas compte de l'interaction entre électrons. Quelle est la longueur caractéristique '0l , et la fonction enveloppe décrivant le mouvement relatif des deux électrons dans l'état fondamental? 4. Si on omet, dans l'équation de la question 2, le potentiel de confinement, définir une seconde longueur caractéristique a. 5. Vérifier que la fonction écrite à la question 3 est normée et calculer, à l'ordre le plus bas d'un calcul de perturbation, l'effet du potentiel d'interaction dans l'état fondamental. 6. Comparer à la figure ci-contre, extraite de
l'article de Lamouche et Fishman.
0
-V0
0 x, y, z
E
Energie des états à deux électrons dans une boîte parabolique (Lamouche et Fishman, J. Phys. Condens. Matter 10 (1998) 7857)

130
7. Tracer les énergies des états fondamentaux avec 0, 1, 2 électrons dans la boîte, en fonction de 0l . On se bornera à tracer les valeurs d'énergies négatives et on commentera qualitativement le comportement attendu pour des boîtes plus petites.
• • • • • •
On rappelle quelques résultats concernant l'oscillateur harmonique à une dimension:
• hamiltonien: 222
22
*21
*2xm
dxd
mω+−
h
• fonctions propres: ( ) ( ) ( )20
20
41
20
2exp/!2
11ll
lxxH
nx nnn −⎟⎟
⎠
⎞⎜⎜⎝
⎛=
πϕ
où ωmhl =0 et où les polynômes de Hermite sont définis par
( ) ( )
( )( )( ) 24
21
2
22
1
0
0
−=
==
⎟⎠⎞
⎜⎝⎛ −=
xxH
xxHxH
xHdxdxxH
n
n
.
• intégrales
( ) 20
2
21exp −
∞ −=−= ∫ p
pp IpdxxxI
20π
=I , 21
1 =I , 42π
=I , 21
3 =I , 8
34
π=I ...
Constantes usuelles:
120
310
1-23
19
34
1085.8
kg 109
JK 1038.1C 106.1
Js 10054.1
−
−
−
−
−
×=
×=
×=
×=
×=
ε
m
ke
B
h
• • • • • • • • • • • • • • •
II. Détermination de l'anisotropie de l'énergie de marches
(tiré de l'article de S.Kodembaka et al, "In-situ high-temperature STM studies of surface dynamics on atomically smooth TiN", Pico 7 (2003) 9, http://www.omicron.de/) La forme d'équilibre d'îlots monoatomiques sur une surface singulière dépend de l'anisotropie de l'énergie des marches (de la même façon que la forme d'équilibre de cristaux dépend de l'énergie de surface). La construction de Wulff permet de tracer la forme d'équilibre à partir du diagramme polaire de l'énergie: l'objet de cette étude est de réaliser le passage inverse, c'est-à-dire de déduire l'anisotropie de l'énergie de marche à partir de l'observation d'îlots. La figure 1 présente des images (200×190 nm) de microscopie tunnel (STM), réalisées sur une surface (001) de TiN (un matériau de structure NaCl). Sur la surface bien préparée, on a

131
déposé 0.15 monocouche de matériau. On a ensuite effectué un recuit à 1123 K pendant une durée tr, et on a enregistré les images STM de la même zone pour trois temps de recuit différents (a) tr = 0, (b) tr = 30 mn, (c) tr = 169 mn. On a également choisi quatre petits îlots (numérotés 1 à 4) et on a tracé en (d) l'aire de ces îlots en fonction du temps de recuit tr: ces îlots disparaissent pendant le recuit. D'autres îlots sont stables: la figure 2 représente deux images (33.8×33.8 nm) successives, pendant recuit, d'un îlot stable. De la stabilité de cet îlot, on déduit qu'il a atteint une forme d'équilibre. Les bords de l'image sont parallèles aux directions <110>.
On prend comme origine O le centre de l'îlot, et on repère la direction d'une marche en surface par l'angle polaire ϕ de la normale à la marche, l'angle ϕ = 0 désignant la direction [110]. On définit l'énergie par unité de longueur de marche β (ϕ). On sera amené à tracer deux types de courbes: - le diagramme polaire de β, c'est à dire la courbe ( )ϕβ obtenue en traçant à partir de O dans la direction ϕ un segment de longueur proportionnelle à β. On notera H le point courant de cette courbe, et ses coordonnées cartésiennes (ξ, η); - la forme de l'îlot, défini par ses coordonnées cartésiennes (x,y), ou en coordonnées polaires (ρ, θ). On notera M le point courant du bord de l'îlot. La construction de Wulff permet de tracer ρ(θ) à partir de β(ϕ). On rappelle qu'il existe deux formes de la construction de Wulff. La première s'applique à des matériaux très anisotropes et n'envisage qu'un nombre fini de directions possibles. La seconde
s'applique à des matériaux à faible anisotropie et s'écrit sous une forme continue:
OM = λ(β uρ + β ' uϕ ), (Eq.1) où uρ est le vecteur unitaire d'angle polaire ϕ, uϕ le vecteur unitaire d'angle polaire ϕ+π/2, et
ϕββ dd=' . Si on veut seulement la forme d'équilibre, on peut poser λ = 1. Si on veut également obtenir l'aire d'un îlot en équilibre pour une sursaturation ∆µ, alors λ = n ∆µ, où n est la densité d'atomes. On peut développer l'équation (Eq.1):
( ) ( ) ( )( ) ( ) ( )ϕβϕβθρ
ϕβϕβθρcos'sinsinsin'coscos
+=−=
a b
c
25 nm
Fig. 1
Fig. 2
a b
α
θ O
M

132
(Eq.2) Cette relation permet de calculer les coordonnées polaires (ρ, θ) du point M associé au point H de coordonnées (β, ϕ). 1. En première approximation, les îlots observés ont la forme d'un carré de côté 2c. Quelle est la direction (exprimée par la valeur de ϕ et par ses indices <lmn>) pour laquelle β est minimale? 2. Quelle condition devrait vérifier β (<100>) pour que les angles des îlots ne soient pas tronqués? 3. Il est visible que les angles sont arrondis, avec un rayon b.
a. Evaluer, pour l'îlot de la figure 2, les longueurs c, b et a=c-b. b. Paramétriser les coordonnées cartésiennes des points d'un "angle arrondi" (on
prendra comme paramètre l'angle polaire α dans le cercle). c. Calculer αθ dd (on en aura besoin à la question 7).
4. Montrer que cette forme d'îlots est la forme d'équilibre si ( ) ( ) ( )[ ]ϕϕββϕβ sincos10 ++= pour 0<ϕ<π/2. 5. Tracer le diagramme polaire en le complétant pour les autres valeurs de ϕ. Déduire alors de la question 3 le rapport β(<110>) / β(<100>). 6. On cherche à établir une relation inverse, qui permettra de déduire β (ϕ) de l'observation d'un îlot ρ (θ) quelle que soit sa forme. En particulier, on va démontrer la relation
22
2
'ρρ
ρβ+
= , où θρρ dd=' (Eq.3)
Pour cela: a. Utiliser (Eq.2) pour exprimer ( )θtan et en déduire ϕθ dd ; b. Différencier chacune des équations de (Eq.2) par rapport à θ et ϕ , et en déduire
une expression de ( )ϕβ
cos1 et ( )ϕβ
sin1 en fonction de 1/ρ, (1/ρ)', θ.
c. En déduire l'expression (Eq.3); Cette expression permet de calculer numériquement ( )ϕβ à partir de l'observation d'un îlot. 7. En restant dans une étude analytique, à partir de la question 3,
a. Calculer β (α) b. Montrer que ϕ = α (par exemple, en explicitant les expressions obtenues à la
question 6b). 8. On observe sur la figure 1 une évolution marquée des petits îlots d'une part, des gros îlots d'autre part. Discuter qualitativement cette évolution (on reprendra l'équation Eq.1 avec λ = n ∆µ, et on calculera le potentiel chimique local au voisinage des îlots, et on en déduira l'évolution de deux îlots voisins).