
Devenir du médicament dans l'organisme : paramètres … · A. Injection directe d'un bolus :...
20
BMCTTM - Devenir du médicament dans l'organisme : paramètres pharmacocinétiques 24/09/2014 CLEMOT Cécile L2 (relecteur : Hamza Berguigua) BMCTTM N. Simon 20 pages Devenir du médicament dans l'organisme : paramètres PK (Paramètres pharmacocinétiques) A. Injection directe d'un bolus : Lorsqu'on choisit de faire un bolus intra-veineux, il y a immédiatement une concentration maximale du produit dans le sang. Le bolus est immédiat et il ne peut y avoir ensuite qu'une décroissance de la concentration du produit. Si l'on veut obtenir une concentration maximale d'emblée, le bolus est obligatoire. 1/20 Plan : A. Injection directe d'un bolus B. Perfusion intra-veineuse C. Voie orale D. Analyse non compartimentale I. Le Tmax et le Cmax II. Demi-vie III. Surface sous la courbe (AUC) IV. Biodisponibilité V. Clairance plasmatique VI. Volume de distribution E. Cas d'une perfusion continue F. Prises orales répétées G. Lien entre la concentration et l'effet (pharmacodynamique)
-
Upload
truongtuong -
Category
Documents
-
view
218 -
download
0
Transcript of Devenir du médicament dans l'organisme : paramètres … · A. Injection directe d'un bolus :...

BMCTTM - Devenir du médicament dans l'organisme : paramètres pharmacocinétiques
24/09/2014CLEMOT Cécile L2 (relecteur : Hamza Berguigua)BMCTTMN. Simon20 pages
Devenir du médicament dans l'organisme : paramètres PK(Paramètres pharmacocinétiques)
A. Injection directe d'un bolus :
Lorsqu'on choisit de faire un bolus intra-veineux, il y a immédiatement une concentration maximale du produit dans le sang. Le bolus est immédiat et il ne peut y avoir ensuite qu'une décroissance de la concentration du produit. Si l'on veut obtenir une concentration maximale d'emblée, le bolus est obligatoire.
1/20
Plan :
A. Injection directe d'un bolus B. Perfusion intra-veineuse C. Voie orale D. Analyse non compartimentale
I. Le Tmax et le Cmax II. Demi-vieIII. Surface sous la courbe (AUC)IV. BiodisponibilitéV. Clairance plasmatiqueVI. Volume de distribution
E. Cas d'une perfusion continue F. Prises orales répétées G. Lien entre la concentration et l'effet (pharmacodynamique)

BMCTTM - Devenir du médicament dans l'organisme : paramètres pharmacocinétiques
Pour 90% des médicaments, le fait de doubler ou diviser les doses augmente ou diminue proportionnellement la concentration (et ainsi la surface sous la courbe): il y a un effet proportionnel, une relation linéaire entre la dose et la concentration.C'est donc une cinétique linéaire même si la courbe de décroissance n'est pas une droite.
B. Perfusion intra veineuse :
Ici, on a plusieurs représentations d'une même administration de 2 grammes, mais avec des durées de perfusions différentes. Plus la perfusion est longue, plus la concentration maximale (Cmax) est basse : la durée de perfusion est donc à prendre en compte.
Cmax survient à la fin de la perfusion : la concentration augmente jusqu'à la fin de la perfusion (en aucun cas il ne peut y avoir de diminution avant la fin de la perfusion). Il est donc très important de préciser la durée de perfusion.
Après la fin de la perfusion, la concentration peut décroître très vite.
– Si le médicament, pour être efficace, doit être en concentration élevée alors on choisira une durée de perfusion courte.
– Si au contraire on veut une une présence plus longue du médicament, alors on augmente la durée de perfusion, mais Cmax diminuera.
La quantité de médicament reste la même donc la surface sous la courbe reste identique, même si on modifie la durée de l'injection (il y aura simplement une déformation de cette surface sous la courbe).
2/20

BMCTTM - Devenir du médicament dans l'organisme : paramètres pharmacocinétiques
C. Voie orale :
Le choix d'administration d'un médicament le plus fréquent est la voie orale.
Quand un patient prend un médicament, la concentration part de zéro. Le médicament passe plusieurs obstacle donc il mettra plus ou moins longtemps avant d'atteindre la concentration maximale (qui n'est pas à un moment précis, contrairement à précédemment où l'on obtenait le pic au moment de l’arrêt de la perfusion).
L'indication est donc plus incertaine, par exemple « une heure + ou – une demi heure » (car cela va dépendre de plusieurs choses).
Si l'on fait varier la dose, dans la majorité des cas, le médicament a une cinétique linéaire (quand on augmente la dose, la concentration augmente proportionnellement).Mais si l'on continue d’augmenter, il y a un risque de saturation : l'organisme ne peut plus éliminer. Il y a alors une accumulation et ce n'est plus linéaire. (C'est donc linéaire pour les doses thérapeutiques.)
On observe également que le temps pour lequel la concentration est maximale (Tmax) reste le même quelque soit la dose. Par exemple, si l'on dose 1g ou 3g, le Tmax reste le même si la cinétique est linéaire.
Donc si la cinétique du médicament est linéaire le Tmax est constant quelque soit la quantité administrée, en revanche la Cmax augmente de façon proportionnelle à la dose, tout comme la surface de la courbe (c'est à dire l'exposition).
3/20

BMCTTM - Devenir du médicament dans l'organisme : paramètres pharmacocinétiques
Attention : Tmax et Cax ne permettent pas de prévoir les effets du médicament mais juste de connaître le devenir du médicament : il s'agit de la pharmacocinétique (effets de l'organisme sur le médicament), et non pas de la pharmacodynamique (effets du médicament sur l'organisme).
Quand on parle de pharmacocinétique, on parle de Tmax (pic de concentration) et souvent la représentation est asymétrique (la montée est plus rapide que la descente pour 90% des médicaments).
Si maintenant on s’intéresse à l'effet du médicament (par exemple sur la fréquence cardiaque, sur la température...) et qu'on le mesure à intervalles réguliers, on observe qu'il y a un effet nul si le patient n'a pas pris le médicament, que l'effet arrive après la prise du médicament, puis que l'effet s’épuise.
Il ne faut pas confondre le pic de concentration et le pic d'effet.Le Tmax correspond au temps nécessaire pour obtenir Cmax et il diffère du temps nécessaire pour obtenir l'effet maximal.
Ce dernier délai peut aller jusqu'à plusieurs semaine ( par exemple un anti dépresseur ne sera réellement efficace qu'au bout de 10 jours) mais peut aussi être quasi inexistant (ce qui est le cas pour les médicaments concernant la fréquence cardiaque).
On a donc une cinétique des concentrations et une cinétique des effets qui ne sont pas parallèles. La notion de Tmax doit être dissociée du temps d'obtention de l'effet maximal même si les deux sont liés.
On ne peut pas prescrire sans connaître la cinétique du médicament.
4/20

BMCTTM - Devenir du médicament dans l'organisme : paramètres pharmacocinétiques
D. Analyse non compartimentale :
I. Le Tmax et le Cmax :
Le Cmax et le Tmax sont des valeurs observées et non pas calculées.Le Tmax est surtout important pour la voie orale. En effet, quand il y a une perfusion c'est le médecin qui choisit le Tmax, alors que par voie orale ça dépend de l'organisme et des propriétés pharmaco-chimiques du médicament.
Si le médicament est récent et que l'on cherche à connaître le Tmax, on fait un test sur un groupe de personnes dont on prélève plusieurs échantillons sanguins.
Sur le graphique ci-dessus, on a effectué 7 prélèvements, on obtient donc 7 concentrations différentes. On observe que la concentration la plus importante correspond au 3eme prélèvement. On a donc un Tmax de une heure, et Cmax égal à 45mg/L.
Mais si on n'avait fait que 6 prélèvements on remarque que le Cmax aurait été de 30mg/L et le Tmax de 2h30.L'idéal serait donc de faire le plus de prélèvements possibles.
Le Cmax et le Tmax déterminés sont donc très dépendants du moment où l'on a effectué le prélèvement mais
5/20
BMCTTM - Devenir du médicament dans l'organisme : paramètres pharmacocinétiques
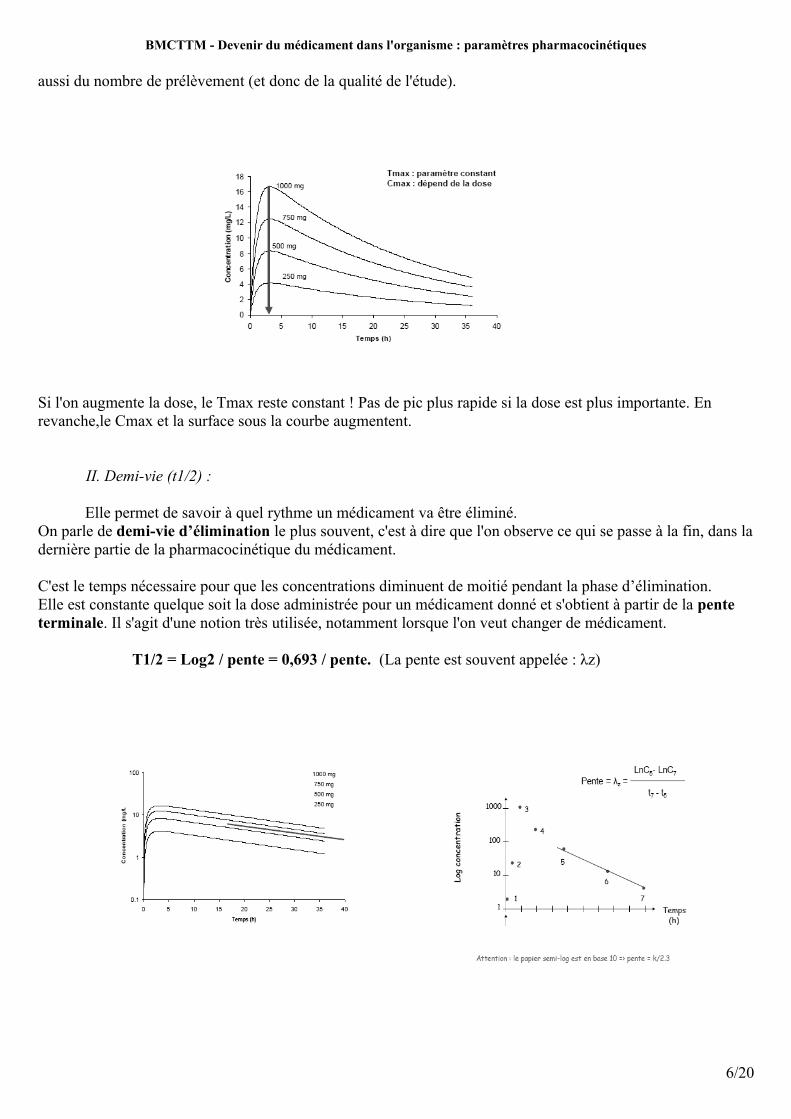
aussi du nombre de prélèvement (et donc de la qualité de l'étude).
Si l'on augmente la dose, le Tmax reste constant ! Pas de pic plus rapide si la dose est plus importante. En revanche,le Cmax et la surface sous la courbe augmentent.
II. Demi-vie (t1/2) :
Elle permet de savoir à quel rythme un médicament va être éliminé.On parle de demi-vie d’élimination le plus souvent, c'est à dire que l'on observe ce qui se passe à la fin, dans la dernière partie de la pharmacocinétique du médicament.
C'est le temps nécessaire pour que les concentrations diminuent de moitié pendant la phase d’élimination. Elle est constante quelque soit la dose administrée pour un médicament donné et s'obtient à partir de la pente terminale. Il s'agit d'une notion très utilisée, notamment lorsque l'on veut changer de médicament.
T1/2 = Log2 / pente = 0,693 / pente. (La pente est souvent appelée : λz)
6/20

BMCTTM - Devenir du médicament dans l'organisme : paramètres pharmacocinétiques
Pour calculer la pente, on peut regarder les logarithmes des concentrations au cours du temps, ce qui permet de linéariser la dernière partie car les cinétiques sont le plus souvent des exponentielles. A partir de cette linéarisation on peut calculer la pente qui est toujours la même, peu importe la dose. Ainsi la demi-vie aussi est constante.
Sur l'image de droite :on a fait 7 prélèvements. Pour calculer la pente d’élimination, c'est facile entre les points 5 et 7. On choisit en règle générale la dernière phase qui commence ici à peu près au point 5.Attention: le log n'a pas d'unité et le temps est en seconde donc la pente sera l'inverse du temps.
C'est utile pour le patient : par exemple si on a un effet indésirable il faut arrêter de prendre le médicament mais le moment où celui-ci sera éliminé va dépendre de la demi-vie (à chaque demi-vie la concentration baisse de moitié).
On accepte classiquement la notion de 5 (ou 7 pour plus de sureté) demi-vies pour dire qu'un médicament a été éliminé (exemple : si la demi-vie est de 4h alors au bout de 20h le patient n'a presque plus de médicament dans l'organisme).
III. Surface sous la courbe (AUC) :
Elle reflète la quantité de médicament ayant atteint la circulation générale, c'est l'exposition du patient au médicament. Son unité est la concentration x temps.
AUC0-tlast : de t0 au dernier prélèvement AUC0-tau : de t0 à la prochaine administration AUC0-∞ : de t0 à l’infini
Quand on avale un médicament, il y a une fraction perdue par le franchissement des différentes barrières (intestinales, hépatiques...) et c'est seulement après le passage du foie que le médicament va dans le cœur, le sang et va être redistribué dans l'organisme. Il faut évaluer les pertes lorsqu’on donne un médicament autrement que par perfusion.
7/20

BMCTTM - Devenir du médicament dans l'organisme : paramètres pharmacocinétiques
On ne peut pas mesurer la dose molécule par molécule mais on sait qu'elle est proportionnelle à la surface sous la courbe.Donc la surface sous la courbe est le reflet de la dose, ( AUC par voie intra-veineuse est la référence par rapport à la dose administrée), mais ce n'est pas la dose.
Si on reprend l'exemple des 7 prélèvements :
On procède par étape : on calcule AUC entre 2 points, par exemple entre le 3eme et le 4eme prélèvement (il s'agit tu trapèze gris qui a la même surface que le rectangle noir).On fait ensuite la somme de tous les rectangles. C'est une méthode approximative mais c'est simple et efficace.Il existe d'autres méthodes plus sophistiquées.
Après le dernier prélèvement on connaît la pente, donc on imagine que la pente reste la même. En divisant la hauteur (=concentration) par la pente on a la surface du triangle gris, ce qui nous permet d'extrapoler.
Quand on augmente la dose, AUC augmente de façon linéaire et proportionnelle ce qui n'est pas toujours évident à cause du phénomène de saturation. Plus généralement lors d'utilisations de « doses thérapeutiques » si on augmente la dose, on augmente l'exposition de façon proportionnelle.
8/20

BMCTTM - Devenir du médicament dans l'organisme : paramètres pharmacocinétiques
IV. Biodisponibilité (F) :
Elle ne reflète rien d'autre que les pertes qui ont lieu lorsqu’on avale un médicament. C'est une fraction, un pourcentage qui dépend de la résorption, du 1er passage hépatique...
Exemple : Parfois, seulement 20% d'un médicament est retrouvé dans la circulation générale ( 80% de perte!).Si on donne 1g d'un médicament de ce type par voie orale, on aura seulement 200 mg dans la circulation générale, alors que par perfusion il y aurait 1 gramme (aucune perte) ! C'est pourquoi il faut toujours connaître la biodisponibilité d'un médicament.
F dépend de la voie d'administration ( suppositoire, crème, collyre, gélule, comprimé, sirop, etc ).On retrouve F dans le Vidal.
Comment estime-t-on ce pourcentage, cette fraction de médicament qui pénètre dans l'organisme ?On compare avec la surface sous la courbe obtenue lors d'une perfusion.
• La biodisponibilité absolue se calcule en référence à la voie intraveineuse.
F = (AUCpo / AUCiv) x (Doseiv / Dosepo )
Si on prend le même médicament mais de 3 types différents :
– en haut a gauche : AUC est à peu près la même alors il n'y a pas de perte.
– En haut au milieu : le pourcentage passe à 50 % (par exemple pour un médicament qui ne supporte pas l'acidité gastrique).
Attention : la galénique d'un médicament est très importante. On ne conserve pas toujours la biodisponibilité du médicament quand on ouvre une gélule ou quand on écrase un comprimé (surtout pour les médicaments gastro-résistants).
– en haut à droite: F=20%
9/20

BMCTTM - Devenir du médicament dans l'organisme : paramètres pharmacocinétiques
• La biodisponibilité relative se calcule en référence à une autre voie d'administration (on peut comparer un médicament et son générique par exemple).
– en bas à gauche : les deux produits ont un profil équivalent mais l'AUC et le pic sont plus faibles pour le médicament bleu (courbe la plus foncée) donc il donne plus de perte que la rouge (courbe la plus claire).
– en bas au milieu : ils sont bio-équivalents ce qui est le cas des médicaments génériques (bio-équivalence entre 0,8 et 1,20)
– en bas à droite : AUC est la même donc ils sont biodisponibles mais ils ne sont pas bioéquivalents car il faut plus de temps pour le comprimé bleu pour être dans l'organisme.
V. Clairance plasmatique :
La clairance c'est un volume divisé par un temps (volume de plasma totalement épuré du médicament par unité de temps).
Vt = vitesse d’élimination du médicament (concentration divisé par le temps) = CL x CtLa vitesse d’élimination dépend de la concentration : si la concentration est élevée alors la vitesse est très rapide, elles sont proportionnelles.
10/20

BMCTTM - Devenir du médicament dans l'organisme : paramètres pharmacocinétiques
Pour calculer la clairance : CL = (F x Dose) / AUC
(on tient compte de la biodisponibilité pour la voie orale, c'est à dire qu'on multiplie par F).
CL = CL hépatique + CL rénale + ...
Quand un patient est malade la capacité à éliminer un médicament peut varier.La CL est constante pour un médicament donné quand on n'est pas malade mais si par exemple on a une infection rénale alors les capacités d’élimination d'un médicament à élimination rénale prépondérante vont diminuer (la CL va augmenter quand le patient guérit).
La CL est donc constante sauf en cas de pathologie (et en cas de maladie elle dépend de la gravité de cette dernière).
Par exemple :On considère une CL de 100L/h (ce qui est élevé) alors pour une certaine dose les concentrations en médicament sont basses, si une pathologie diminue la CL à 25L/h les concentrations seront donc nettement plus importantes.
11/20

BMCTTM - Devenir du médicament dans l'organisme : paramètres pharmacocinétiques
Les gens avec une fonction rénale normale sont à droite, au delà de 80.
Plus CLCR est basse plus il y a une pathologie grave, puisque plus la fonction rénale est basse plus la CL est basse : il y a une véritable relation entre les 2.
Il faut donc tenir compte des capacités du patient à éliminer une dose et quand on prescrit, on a besoin de savoir comment se comportent ses fonctions d'élimination (rénale, hépatiques...).
Dans l'exemple de cet antiviral (ganciclovir) il faut connaître l'état rénal du patient !
Ici le patient avec un rond noir a une fonction rénale normale mais pour les autres patients elle est perturbée.On ne peut pas prescrire un médicament éliminé par le rein sans se poser de question sur la physiologie du rein.
VI. Volume de distribution (Vd) :
C'est le volume dans lequel devrait se répartir le médicament pour être à la même concentration que dans le plasma.
12/20

BMCTTM - Devenir du médicament dans l'organisme : paramètres pharmacocinétiques
Il s'agit d'un volume virtuel qui est utilisé pour comparer des médicaments, mais qui n'a aucune traduction physiologique. Il reflète si le médicament a la capacité de sortir du sang.
Le fait d’être dans le sang n'est pas suffisant, le médicament doit par exemple pouvoir passer dans le muscle ou dans le cerveau. Donc il doit s'extraire du sang pour aller vers sa cible, ce qui est reflété par sa capacité à se distribuer dans l'organisme.
Le Vd nous dit seulement si le médicament sort du compartiment sanguin (avec un ordre de grandeur).
Plus le Vd est élevé, plus la distribution tissulaire est importante. Le plus petit Vd est 0.06 L/kg et il n'y a pas de limite supérieure.Le calcul est basé sur la pente terminale (λz) : méthode rudimentaire mais efficace. Vd = CL / λz
Cette méthode est utilisée dans les analyses non compartimentales. Elle nécessite une bonne estimation de la pente.
Le Vd est uniquement utilisé en recherche.
Exemple : - Un médicament est très hydrophile, il va aller se distribuer partout où il y a de l'eau.Mais si on a une insuffisance cardiaque et que l'estomac se remplit d'eau (ascite) le médicament va aller dans les œdèmes, ce qui a des conséquences sur la quantité de médicament capable d'atteindre la cible.
- Pour un médicament lipophile. S'il n'y a pas beaucoup de gras dans l'organisme (sportif), la
distribution va être différente par rapport à une personne dont la masse adipeuse est plus importante. Donc on ne va pas donner la même dose à 2 personnes qui n'ont pas la même masse de graisse.
Plus le Vd est important, moins il y a de médicament dans le sang. Le Vd joue beaucoup sur le pic de concentration (si Vd est faible le pic sera plus important).
t1/2 = 0.693 / λz et CL = λz x Vd
D ’où: t1/2 = (0.693 Vd) / CL
13/20

BMCTTM - Devenir du médicament dans l'organisme : paramètres pharmacocinétiques
La demi-vie dépend de 2 choses :– de la capacité du médicament à se distribuer (Vd).– de la clairance.
Plus CL est importante plus le médicament est éliminé rapidement. Donc la demi-vie est un paramètre intermédiaire, qui dépend d'autres paramètres.
Piège : face à un patient dont la fonction rénale est altéré, la clairance a baissé mais la demi-vie on ne sait pas, car elle dépend aussi de Vd. En général les 2 paramètres sont modifiés. Il faut toujours prendre en compte la CL ET le Vd.
E. Cas d'une perfusion continue :
Le médicament est perfusé à débit constant : R0 Au bout d’un certain temps un plateau (qui reflète l'état d'équilibre) est atteint.
2 paramètres caractérisent le plateau: - la « hauteur ».- le temps pour l’atteindre.
Il faut toujours préciser la dose ET la durée de la perfusion.
Tant que la perfusion est continue, on a un plateau et ensuite une décroissance. Si on attend suffisamment longtemps on obtient un état stationnaire : le débit qui entre est égal au débit de sortie du médicament.
Le débit d'entrée est la dose que divise la durée de perfusion.Le débit de sortie est CL x la concentration
Entrée : R0 = Dose / durée de la perfusion Sortie : Vt = CL x Céq.
Au moment du plateau : R0=CL x Ceq, donc Ceq = Ro / CL.Céq. dépend du débit de perfusion et de la clairance (+++).
14/20

BMCTTM - Devenir du médicament dans l'organisme : paramètres pharmacocinétiques
La clairance est indépendante de notre volonté contrairement à Ro. Donc il faut savoir la valeur de la clairance du patient (qui dépend du patient selon ses pathologie de foie ou de rein) pour obtenir Ceq.
Le temps pour atteindre le plateau ne dépend que de la demi-vie (elle permet de calculer le temps nécessaire pour arriver a l’état d’équilibre) : il faut 5 à 7 demi-vies pour être au plateau (après 5 demi-vies, la concentration atteinte représente 97% de celle obtenue au plateau).
La montée des concentrations est l’image en miroir de la décroissance à l’arrêt de la perfusion.
Ceci est vrai pour la perfusion mais aussi pour la voie orale. Pour juger de l'effet d'un médicament il vaut mieux être à l'état d’équilibre (= stationnaire) car sinon il n'est pas complètement dans l'organisme.
Or la demi-vie peut être longue donc le temps pour atteindre le plateau peut être long.Par exemple pour les antibiotiques dont l'effet dépend de le concentration, si le médicament met du temps à atteindre le plateau alors il y a une perte de chance pour le patient !
Pour atteindre d'emblée la concentration d'équilibre on fait 2 administrations en même temps : on démarre la perfusion et on injecte un bolus (dose de charge).
Dose = Céq x Vd
Les concentrations s'additionnent et on obtient directement un plateau. Attention : il faut démarrer perfusion et bolus en même temps, sinon il n'y a aucun intérêt.
15/20

BMCTTM - Devenir du médicament dans l'organisme : paramètres pharmacocinétiques
F. Administrations orales répétées :
Si on prescrit un médicament 3 fois par jour par exemple à 8h, 12h ( le médicament n'a pas eu le temps de diminuer donc il y a sommation) et a 20h.
Il y a succession de pics, ce n'est pas très homogène. Si le patient prend le médicament 3 jours de suite, on a une fluctuation et il va falloir attendre 5 demi-vies après avoir démarré le traitement pour que la fluctuation soit constante.
Les concentrations maximale (midi) et minimale (matin) sont constantes.
On a une oscillation/fluctuation stationnaire des concentrations (pas de plateau).
Cette fluctuation est importante car on veut que les concentrations soit toujours dans une zone d'activité, tout en évitant que les concentrations soit trop hautes (risque de toxicité).
Que ce soit la première ou la 5ème administration, comme la dose est toujours la même la surface sous la courbe reste la même.Les surface hachurées sont égales même si les formes sont différentes.
Si on veut on peut calculer le pourcentage de fluctuation.
16/20

BMCTTM - Devenir du médicament dans l'organisme : paramètres pharmacocinétiques
Le temps pour atteindre l'équilibre dépend de la demi-vie du médicament.Pour la voie orale le temps pour atteindre l’équilibre est de 5 demi-vies.
Ration d’accumulation : r apport des AUC0-t entre J1 et Jn.
Si la demi-vie est courte, les fluctuations se stabilisent vite, ce qui n'est pas le cas si la demi-vie est longue.
Il faut attendre que les fluctuations se stabilisent pour voir l'effet d'un médicament. Par exemple, si la demi-vie est 24h on doit attendre 5 jours pour avoir un effet stable ! Si au bout de 5 jours ce n'est pas suffisant alors on augmente la posologie et il faut attendre encore 5 jours pour évaluer les effets de la posologie.
C'est important quand on veut juger de l’efficacité d'une dose.
G. Lien entre la concentration et l'effet (pharmacodynamique) :
Les deux cinétiques ne sont pas superposables (effet/concentration).
17/20

BMCTTM - Devenir du médicament dans l'organisme : paramètres pharmacocinétiques
Dans un cas théorique quand la concentration augmente, l'effet augmente et si on baisse la concentration l'effet va baisser donc on a une relation linéaire.Ce cas de figure existe par exemple pour les médicaments qui ont un effet sur la fréquence cardiaque.
Dans beaucoup d'autres cas, quand la concentration augmente, l'effet atteint un effet maximal.
Par exemple, si on prend un médicament pour la fièvre : plus on prend de médicament, plus la fièvre baisse mais on ne se retrouve pas avec une température de 30 degrés pour autant car il y a un effet maximal.
Pour certains médicament l'effet augmente linéairement, mais souvent la relation est plutôt sigmoïdale avec l'obtention d'un effet maximal.
18/20

BMCTTM - Devenir du médicament dans l'organisme : paramètres pharmacocinétiques
Conclusion :
Les optimisations de posologies doivent se baser sur des études pharmacocinétiques ET pharmacodynamiques.
Ces paramètres étudiés peuvent être utilisés au lit du patient.Ces méthodes d'analyses non compartimentales sont des analyses rudimentaires mais ce sont des techniques robustes.
Ce sont des notions simples mais il existe des méthodes plus sophistiquées comme la régression non-linéaire à effets mixtes (nomem), il s’agit de trouver deux fonctions mathématiques :
– une décrivant l’évolution des concentrations (surrogate marker) en fonction de la dose (PK)
– une décrivant l’évolution de la réponse clinique en fonction des concentrations (PD)
Par exemple : comme pour un simulateur aérien (ordinateur avec des équations), en pharmacocinétique on a des modèles mathématiques qui vont décrire l’évolution des concentrations donc on peut faire des simulations.
Ce n'est pas seulement de la recherche ! Les simulateurs sont utilisés en anesthésie notamment. On a un ordinateur qui est branché à un pousse seringue électrique et l’anesthésiste précise la concentration voulue et l'ordinateur va calculer pour obtenir un débit qui aille le plus vite possible.
Beaucoup de produits sont ainsi directement pilotés.
Cela s'utilise en routine et notamment au bloc ce qui permet d’éviter le surdosage et de ne pas gaspiller les médicaments.
19/20

BMCTTM - Devenir du médicament dans l'organisme : paramètres pharmacocinétiques
20/20


















