
desdispositifs médicaux selonladirective 93/42/CEE marquage_CE.pdf · 14 GuidedumarquageCE...
52
1 Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE
Transcript of desdispositifs médicaux selonladirective 93/42/CEE marquage_CE.pdf · 14 GuidedumarquageCE...

1
Guidedumarquage CE
des dispositifsmédicaux
selon la directive93/42/CEE

3Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE2
EDITORIAL
Aucours de la «TraverséeduDésert » des entreprises, longprocessusqui chemine depuis les laboratoires et les prototypes jusqu’aux dispositifsmé-dicaux commercialisables à destination des acteurs du système de santé, lemarquageCE est la seule étape réellement obligatoire, et donc absolument in-contournable. Il surseoit auxmécanismesde remboursement, eux-mêmessou-mis aux exigences de démonstration de service médical. (Voir étude « TdD » :www.utc.fr/agbm.)
C’est aussi aujourd’hui un des rares outils de régulation pour lestechnologies de la santé dans le système public, qui dépasse les frontièresdes états. Le marquage CE est très probablement appelé à se renforcer, à seperfectionner dans les années à venir, à élargir ses prérogatives, en particulieravec des exigences cliniques minimales, et à homogénéiser les efforts desacteurs épars.
Il était important de réaliser un document de synthèse pour permet-tre à tous ceux qui sont au cœur de l’innovation, en particulier ceux qui conçoi-vent les dispositifsmédicaux, d’êtremieux éclairés : terminologie, démarche,dossier, certification, etc. C’est donc avec grand plaisir que l’AGBM s’associeà la diffusion de ce document précieux et espère sa réussite.
Compte tenu de la conjonction des besoins grandissants liés au vieil-lissement de la population et du potentiel de croissance avec, par exemple,l’inflexion prévisible due aux technologies de l’information et de la communi-cation, le secteur des technologies de santé, aumême titre que ceux de la dé-fense, du transport ou de l’énergie, devrait être stratégique. C’est d’une bonnecompréhension des mécanismes auxquels sont confrontés ceux qui entre-prennent et contribuent à la création de richesse, mais aussi de la créationou l’amplification du marché de santé publique par la demande médicale etl’assimilation adéquate par la clinique, que les progrès peuvent venir.
François LANGEVINPrésident de l’AGBM

5Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE4
1 - INTRODUCTION
Ce guide s’adresse à tous les industriels dumonde des dispositifsmédicaux*1
qu’ils soient fabricants*, sous-traitants, fournisseurs ou distributeurs. Il estdestiné à éclairer ces industriels sur la notion de « marquage CE ».
Le marquage CE* est un passeport communautaire obligatoire pour tous lesdispositifsmédicauxmis sur lemarché de l’Union Européenne. Ce guide per-met de replacer le marquage CE dans un contexte historique, économique etpolitique et de présenter lesmoyens pouvant êtremis enœuvre pourmarquerCE des dispositifs médicaux.
L’industriel concerné par le marquage CE de dispositifs médicaux va devoirfaire des choix stratégiques et techniques. Ce guide permettra de l’orienterafin de prendre les décisions s’imposant en fonction de l’état d’organisation deson entreprise, de l’avancement dans son projet de recherche et développe-ment et de la conformité de son produit fini à des normes* référencées. Plusla notion de marquage CE sera intégrée tôt dans le développement d’un dis-positif médical, plus la réalisation de ce marquage CE sera facilitée.
Le marquage CE est effectué sous la responsabilité du fabricant avec l’inter-vention éventuelle d’un organisme notifié* en fonction du niveau de risque duproduit. Différentes méthodes de marquage CE sont proposées au fabricant.Le choix de la méthode de marquage CE est important car celui-ci entraînedes contraintes en termes de coût et de délai non négligeables. C’est pour ac-compagner les industriels du secteur des dispositifsmédicaux à faire lemeil-leur choix pour la pérennité de leur entreprise et le succès de la mise sur lemarché de leurs dispositifs médicaux que le CRITT Santé Bretagne a édité ceguide.
Ce guide dumarquage CE des dispositifsmédicaux a été construit demanièreà représenter le cheminement à suivre pour obtenir cemarquage. Il ne permetpas, à lui seul, de réaliser le marquage CE car il ne saurait se substituer auxnormes et réglementations en vigueur. Par contre, il donne les clés essen-tielles pour comprendre le marquage CE, trouver les normes et réglementa-tions applicables, réaliser le dossier technique* demarquage CE etmettre enplace un système de management de la qualité*.Ce guide a été rédigé par Mlle Cynthia Cottereau, Twoksa Conseil,
Conseil en Qualité et Affaires Réglementaires des Dispositifs Médicaux.
1Les mots suivis d’un astérisque (*)sont présents dans le glossaire à la fin du guide

7Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE6
2 - TABLE DES MATIÈRES
1 Introduction 5
2 Table des matières 7
3 Qu’est-ce que le marquage CE ? 11
3.1 Définition de la « nouvelle approche » 11
3.1.1 Le contexte historique 11
3.1.2 But de la nouvelle approche 12
3.1.3 Principe de la nouvelle approche 14
3.1.4 Relations entre directives et normes 14
3.2 Définition du marquage CE 17
4 Les directives concernant les dispositifs médicaux 19
4.1 Définitions 19
4.1.1 Dispositif Médical ou DM 19
4.1.2 Accessoire 20
4.1.3 Dispositif Médical de Diagnostic in vitro ou DMDIV 20
4.1.4 Dispositif Médical Actif ou DMA 21
4.1.5 Dispositif Médical Implantable Actif ou DMIA 21
4.2 Les directives applicables en fonction du typede dispositif médical 22
4.2.1 Les DM, DMDIV, DMA, DMIA 22
4.2.2 Les DM incorporant un médicament ou « produits frontières » 23
4.3 La mise à jour des directives 93/42/CEE, 90/385/CEE et 98/8/CE 25
4.3.1 Les modifications apportées par la directive 2007/47/CE 25
4.3.1.1 Les logiciels 25
4.3.1.2 L’évaluation clinique 25
4.3.1.3 Relations avec l’organisme notifié 26
4.3.1.4 Le management des risques 27
4.3.1.5 La classification 27
4.3.2 La transposition des directives 28
4.3.3 La période transitoire 28
5 Les acteurs du marquage CE 31
5.1 La Commission Européenne 31
5.2 Les Autorités Compétentes 31

9Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE8
8.1.4.3 Conception et développement (§7.3) 69
8.1.4.4 Achats (§7.4) 70
8.1.4.5 Production et préparation du service (§7.5) 70
8.1.4.6 Maîtrise des dispositifs de surveillance et de mesure (§7.6) 71
8.1.5 Mesures, analyse et amélioration (§8) 72
8.2 Annexe V : Assurance de la qualité de la production 73
8.3 Annexe VI : Assurance de la qualité des produits 73
9 Les annexes III, IV et VIII 75
9.1 Annexe III de la directive 93/42/CEE 75
9.2 Annexe IV de la directive 93/42/CEE 75
9.3 Les dispositifs médicaux sur-mesure 76
9.4 Les DM destinés à des investigations cliniques 77
9.5 Les DM de classe Is ou Im 78
10 Les aspects réglementaires à ne pas oublier 79
10.1 La matériovigilance 79
10.2 La Post-Market surveillance 79
10.3 La loi anti-cadeaux 80
11 Glossaire 81
12 Table des tableaux 93
13 Table des figures 93
14 Annexe 1 : La directive 93/42/CEE 95
15 Annexe 2 : Pour plus d’information… 103
5.3 Les Organismes Notifiés 32
5.4 Le Fabricant 34
5.5 Le Mandataire dans l’Union Européenne 35
5.6 Les Distributeurs et Agents 36
5.7 Les Sous-traitants et Fournisseurs 36
6 La démarche de marquage CE 39
6.1 Mon produit est-il un dispositif médical ? 39
6.2 Dans quelle classe de risques mon dispositif se situe-t-il ? 40
6.3 Quel mode de marquage CE vais-je choisir ? 46
6.4 Quel organisme notifié vais-je choisir ? 49
6.5 Planification d’une démarche de marquage CE 49
7 Le dossier technique de marquage CE 51
7.1 Les exigences essentielles de la directive 93/42/CEE 51
7.2 Les parties principales du Dossier Technique de Marquage CE 54
7.2.1 Identification du fabricant et de son représentant 54
7.2.2 Identification du dispositif médical et de ses variantes 54
7.2.3 Dossier de recherche et développement 55
7.2.4 Dossier de fabrication 56
7.2.5 Documents d’accompagnement 56
7.2.6 Liste des normes et référentiels revendiqués 57
7.2.7 Analyse de la biocompatibilité 57
7.2.8 Etudes et essais techniques 59
7.2.9 Revue des données cliniques 60
7.2.10 Analyse des risques 60
7.2.11 Suivi après mise sur le marché 62
7.2.12 Revue de conformité aux exigences essentielles 62
7.3 La constitution du dossier technique de marquage CE 64
8 La certification d’entreprise 65
8.1 Annexe II de la directive 93/42/CEE ou l’ISO 13485 65
8.1.1 Système de Management de la qualité (§4) 66
8.1.2 Responsabilité de la direction (§5) 67
8.1.3 Management des ressources (§6) 68
8.1.4 Réalisation du produit (§7) 68
8.1.4.1 Planification de la réalisation du produit (§7.1) 68
8.1.4.2 Processus relatifs aux clients (§7.2) 69

11Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE10
3 - QU’EST-CE QUE LE MARQUAGE CE ?
3.1 Définition de la « nouvelle approche »
3.1.1 Le contexte historique
Née dans les années 50 avec l’objectif de mettre fin aux guerres qui ontrégulièrement secoué le continent, l'Union Européenne, issue de laCECA2, unit progressivement les pays européens sur le plan économiqueet politique. Dès 1957, le traité de Rome institue la Communauté Econo-mique Européenne (CEE) également appelée «marché commun ». Dansles années 60, l'économie européenne connait une période faste grâce àl'abandon de l'imposition des droits de douane par les pays membres dela CEE dans leurs échanges commerciaux. C’est également durant cettedécennie qu’apparaît la politique agricole commune qui a pour objectifd’assurer une production agricole suffisante pour l’ensemble des paysde la CEE.
La CEE continue son élargissement avec l’intégration, entre 1973 et 1986,du Danemark, de l'Irlande, du Royaume-Uni, de la Grèce puis de l’Es-pagne et du Portugal. Durant ces années, les dernières dictatures euro-péennes disparaissent (renversement du régime de Salazar au Portugalet mort du général Franco en Espagne) et la Communauté Européennecommence à intervenir dans les affaires d’état en transférant dessommes considérables dans les régions les plus démunies afin de créerdes emplois et des infrastructures.
C'est en 1986 que l'Acte Unique Européen est signé. Ce traité sert debase à un vaste programme de six ans destiné à supprimer les entravesà la libre circulation des marchandises au sein de la CEE, donnant nais-sance au «marché unique». Le marché unique est achevé en 1993, avecla mise en place des « quatre libertés » :
• Libre circulation des biens
• Libre circulation des services
• Libre circulation des personnes
• Libre circulation des capitaux.
2CECA : Communauté Européenne du Charbon et de l'Acier fondéepar la Belgique, la France, l'Allemagne, l'Italie, le Luxembourg
et les Pays-Bas en 1950.

13Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE12
La Communauté Economique Européenne devient l’Union Européenneen 1993 suite à la signature du traité de Maastricht.
3.1.2 But de la nouvelle approche
La nouvelle approche* est basée sur 3 piliers fondamentaux :
• La résolution du conseil du 7 mai 1985, dans laquelle une nouvelleapproche d’harmonisation technique est vue comme une conditionnécessaire pour assurer la compétitivité de l’industrie européenne.
• La résolution du conseil du 21 décembre 1989 concernant uneapprocheglobale de certificationet de tests, qui pose lesprincipespourune réglementation communautaire de la vérification de conformité.
• La décision du conseil 93/465/CEE qui complète la notiond’approche globale de la résolution citée précédemment. Cettedécision donne les guides généraux et les procédures détailléespour la vérification de conformité qui doivent être utilisés pourles directives* « nouvelle approche ».
La « nouvelle approche » est donc née de la volonté de l’Union Euro-péenne d’harmoniser la réglementation des biens et des marchandisesafin d’assurer leur libre circulation sur le marché intérieur européen etd’accroitre la compétitivité des industries européennes.
L’objectif ultime de cette nouvelle approche est de remplacer les régle-mentations nationales des états membres par une réglementationeuropéennereconnuepar tous, assurant ainsi desexigencesdeperformanceet de sécurité similaires pour les biens et marchandises quel que soitl’état membre dans lequel ils ont été fabriqués. Lemarquage CE est issude la nouvelle approche : tout bien ou marchandise répondant auxexigences énoncées dans les directives « nouvelle approche » doit porterle marquage CE et aucun état membre de l’Union Européenne ne peuts’opposer à la libre circulation de ces produits sur son territoire.
Depuis 1987, 22 directives ont été adoptées sur le principe de la nouvelleapproche (voir Tableau 1). Ces 22 directives ont le double objectifd’assurer la libre circulation des marchandises par une harmonisationtechnique de la réglementation des produits d’un secteur et de garantirun haut niveau de protection du public.
Identifiantde la directive Produit concerné
87/404/CEE Récipients à pression simple
88/378/CEE Jouets
89/106/CEE Produits de construction
2004/108/CE Compatibilité électromagnétique (CEM)
98/37/CE Machines
89/686/CEE Equipements de protection individuelle (EPI)
90/384/CEE Instruments de pesage à fonctionnementnon automatique
90/385/CEE Dispositifs médicaux implantables actifs* (DMIA)
90/396/CEE Appareils à gaz
92/42/CE Chaudières à eau chaude
93/15/CEE Explosifs à usage civil
93/42/CEE Dispositifs médicaux (DM)
94/9/CE Appareils et systèmes de protection destinés à êtreutilisés en atmosphère explosive (ATEX)
94/25/CE Bateaux de plaisance
94/62/CE Emballages et déchets d'emballages
95/16/CE Ascenseurs
97/23/CE Equipements sous pression
98/79/CE Dispositifs médicaux de diagnostic in vitro* (DMDIV)
1999/5/CE Equipements terminaux de télécommunication
2000/9/CE Installations à câbles transportant des personnes
2004/22/CE Instruments de mesure
2006/95/CE Equipements basse tension
Tableau 1 - Liste des directives « nouvelle approche »

15Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE14
3.1.3 Principe de la nouvelle approcheLa particularité de la nouvelle approche réside dans l’innovation de laméthode choisie pour réaliser l’harmonisation au niveau européen (d’oùsa dénomination de « nouvelle approche »). Les directives d’harmonisa-tion « nouvelle approche » sont rédigées selon le principe des « Exi-gences essentielles* ». Chaque directive énonce les exigencesessentielles de performance, de sécurité, de santé et de protection del’environnement auxquelles doivent répondre les produitsmis sur lemar-ché pour pouvoir bénéficier de la liberté de circulation. Les exigencesessentielles sont énoncées de manière générale, les directives ne fixantplus de spécifications techniques précises, produit par produit, commecela se faisait auparavant, dans « l’ancienne approche ».
Les directives « nouvelle approche » définissent également lesmodalitésd’apposition dumarquage CE sur un produit, qui va dépendre des risquesliés au produit. Plus un produit est potentiellement dangereux pour l’uti-lisateur ou un tiers, plus la procédure de vérification de la conformité auxexigences essentielles sera complexe. Le changement notable de la nou-velle approche réside dans le fait que les industriels ont désormais lechoix des moyens pour répondre aux exigences essentielles.
3.1.4 Relations entre directives et normesAvec la nouvelle approche, l’Union Européenne délègue aux organismesde normalisation européens (CEN3 et CENELEC4, notamment) la tâchede définir les spécifications techniques permettant de répondre auxexigences essentielles : les normes.
Lorsque l’application d’une norme permet de répondre aux exigences es-sentielles d’une directive donnée, cette norme est appelée « norme har-monisée ». La liste des normes harmonisées* pour chaque directive estmise à jour et publiée régulièrement au JOUE5 et est accessible sur In-ternet à l’adresse :
http://www.newapproach.org/Directives/DirectiveList.asp.
A titre d’exemple, il existe, à ce jour, 254 normes harmonisées pour ladirective 93/42/CEE relative aux dispositifs médicaux.
L’application des normes harmonisées pour réaliser le marquage CEd’un produit est facultative. Il appartient à l’industriel de choisir lesmoyens les plus appropriés pour répondre aux exigences essentielles dela (des) directive(s) à laquelle (auxquelles) sont soumis ses produits :
• S’il choisit d’appliquer les normes harmonisées, il bénéficiera d’uneprésomption de conformité aux exigences essentielles et l’obtentiondu marquage CE pour le produit considéré sera simplifié.
• S’il choisit de ne pas appliquer les normes harmonisées, l’industrieldevra démontrer par tous moyens la conformité de son produit auxexigences essentielles afin de pouvoir y apposer le marquage CE.
Il existe deux types de norme harmonisée :
• Les normes horizontales* : ce sont des normes générales quis’adressent à l’ensemble des produits d’un domaine. Ce sont, parexemple, des normes demanagement de la qualité ou de la sécuritéou des normes d’essais techniques.
• Les normes verticales* : ce sont des normes spécifiques quis’adressent à un produit particulier ou à une catégorie de produits,définissant des exigences particulières pour ce produit ou cettecatégorie de produit et lesméthodes d’essais permettant demesurerla conformité à ces exigences particulières.
3CEN : Comité Européen de Normalisation4CENELEC : Comité Européen de Normalisation Electrotechnique5JOUE : Journal Officiel de l’Union Européenne

17Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE16
NF EN ISO 14971 - Application de la gestion des risques aux dispositifs médicaux
NF EN ISO 10993 - Évaluation biologique des dispositifs médicaux
NF EN 980 - Symboles graphiques utilisés pour l'étiquetage des dispositifs médicaux
NF EN 1041 - Informations fournies par le fabricant avec les dispositifs médicaux
GANTS MEDICAUX APPAREIL DERADIODIAGNOSTIC ART DENTAIRE
NF EN 455Gants médicauxnon réutilisables
NF EN ISO 21171Gants à usage médical -
Détermination dela poudre résiduelle
en surface
NF EN 60601-1Exigences générales pour
la sécurité de baseet les performances
essentielles
NF EN 60601-1-3Règles générales pourla radioprotection dans
les équipements àrayonnement Xde diagnostic
NF EN 60601-1-6Aptitude à l'utilisation
NF EN 62220-1Caractéristiques desappareils d'imagerieà rayonnement X -Détermination de
l'efficacité quantiquede détection
NF EN 1639Dispositifs médicauxpour l'art dentaire -
Instruments
NF EN 1640Dispositifs médicauxpour l'art dentaire -
Matériel
NF EN 1641Dispositifs médicauxpour l'art dentaire -
Produits
NF EN 1642Dispositifs médicauxpour l'art dentaire -Implants dentaires
NORMES VERTICALES
NO
RM
ESH
OR
IZO
NTA
LES
Figure 1 :Schématisation de la notion de « norme verticale » et de « norme horizontale »
3.2 Définition du marquage CE
Le marquage CE est un symbole apposé sur un produit garantissant quecelui-ci satisfait aux exigences essentielles des directives européennes dontil relève. Ce symbole, signifiant « Communauté Européenne » autorise lalibre circulation du produit dans les états membres de l’Union Européenne.Ceci s’applique aux dispositifs médicaux mais aussi à tous les produitsrelevant des directives « nouvelle approche ».
Le marquage CE désigne également la procédure suivie par un fabricantpour obtenir le droit d’apposer le symbole CE sur son produit.
Le marquage CE est obligatoire pour tous les dispositifs médicaux mis surle marché, excepté les dispositifs sur-mesure* et ceux destinés aux inves-tigations cliniques* qui font l’objet d’une procédure spécifique n’aboutissantpas au marquage CE.
Attention :
• Le marquage CE n’est pas une marque
• Le marquage CE n’est pas une garantie de qualité supplémentaire
- Le marquage CE garantit que le dispositif satisfait aux exigencesessentielles en terme de performance et sécurité. En aucun cas, unfabricant ne peut revendiquer une qualité supérieure de son produitsous prétexte qu’il porte le marquage CE. Tous les dispositifsmédicaux portent ce marquage qui est obligatoire.
• Le marquage CE n’est pas une certification d’entreprise*
- Si la certification d’entreprise peut être nécessaire pour obtenir lemarquage CE, l’apposition du marquage CE n’implique pas forcé-ment que l’entreprise est certifiée.
- La certification qualité d’une entreprise (certificat qualité « ISO »)est une démarche volontaire de la part de l’industriel qui se réaliseen plus du marquage CE (certificat qualité « CE ») bien que lesnormes à appliquer (ISO 13485* pour les dispositifs médicaux) et lesorganismes délivrant les certificats soient les mêmes.

19Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE18
• Le marquage CE n’est pas une certification de produit* (type NF, BS,DIN…)
- La certification de produit selon des normes spécifiques constitueégalement une démarche volontaire de l’entreprise en plus dumar-quage CE. Ces certifications se font auprès d’organismes nationauxet garantissent une qualité du produit. Si le produit possède unecertification produit, le symbole de cette certification viendra en plusdu marquage CE sur le produit.
- Une certification produit n’est en aucun cas suffisante pour obtenirle marquage CE. Elle ne peut pas non plus s’y substituer.
• Le marquage CE n’est pas une homologation
- L’homologation est une ancienne procédure nationale française quipermettait d’obtenir une autorisation de mise sur le marché pourdes dispositifs médicaux spécifiques. Cette homologation a disparule 14 juin 1998, date à laquelle le marquage CE est devenu obliga-toire pour tous les dispositifs médicaux.
• Le marquage CE n’est pas un critère de remboursement
- Un dispositif médical doit être marqué CE pour être mis sur le mar-ché mais celui-ci ne suffit pas pour que le dispositif soit remboursépar la sécurité sociale des pays européens qui en possèdent une.
- Chaque pays européen possède une législation différente en termesde sécurité sociale et de remboursement des dispositifs médicaux.Il convient de réaliser une demande de remboursement danschacun des pays concernés une fois le marquage CE réalisé.
- Si le marquage CE permet la libre circulation dans l’Union Euro-péenne, la procédure de demande de remboursement reste régle-mentée au niveau national pour chacun des états membres.
4 - LES DIRECTIVES CONCERNANTLES DISPOSITIFS MÉDICAUX
4.1 Définitions4.1.1 Dispositif Médical ou DMDéfinition de la directive 93/42/CEE :
« Tout instrument, appareil, équipement, logiciel, matière ou autre arti-cle, utilisé seul ou en association, y compris le logiciel destiné par lefabricant à être utilisé spécifiquement à des fins diagnostic et/outhérapeutique, et nécessaire au bon fonctionnement de celui-ci, destinépar le fabricant à être utilisé chez l'homme à des fins :
• de diagnostic, de prévention, de contrôle, de traitement ou d'atté-nuation d'une maladie,
• de diagnostic, de contrôle, de traitement, d'atténuation ou de com-pensation d'une blessure ou d'un handicap,
• d'étude ou de remplacement ou modification de l'anatomie ou d'unprocessus physiologique,
• de maîtrise de la conception,
et dont l'action principale voulue dans ou sur le corps humain n'est pasobtenue par des moyens pharmacologiques ou immunologiques ni parmétabolisme,mais dont la fonction peut être assistée par de telsmoyens.»
Exemples de dispositifs médicaux :
• Gant médical
• Champ opératoire
• Seringue
• Thermomètre
• Implants et prothèses
• Equipement de radiologie
• Appareil dentaire
• Lunettes correctrices
• Préservatifs
• Logiciel d’aide au diagnostic
• Ceinture lombaire

21Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE20
4.1.2 Accessoire
Définition de la directive 93/42/CEE :
« Tout article qui, bien que n'étant pas un dispositif, est destiné spécifi-quement par son fabricant à être utilisé avec un dispositif pour permettrel'utilisation dudit dispositif conformément aux intentions du fabricant dece dispositif. »
Les accessoires* sont marqués CE séparément du dispositif médicalavec lesquels ils doivent être utilisés. Ils ne relèvent pas obligatoirementde lamême classe, excepté pour les logiciels intervenant sur le fonction-nement ou les performances d’un dispositif médical.
Exemples d’accessoires :
• Produit de nettoyage et désinfection pour lentilles de contactsouples
• Produit lubrifiant et désinfectant pour endoscope
• Logiciel de commande d’un équipement IRM
4.1.3 Dispositif Médical de Diagnostic in vitro ou DMDIV
Définition de la directive 98/79/CE :
« Tout dispositif médical qui consiste en un réactif, un produit réactif, unmatériau d'étalonnage, un matériau de contrôle, une trousse, un instru-ment, un appareil, un équipement ou un système, utilisé seul ou en com-binaison, destiné par le fabricant à être utilisé in vitro dans l'examend'échantillons provenant du corps humain, y compris les dons de sang etde tissus, uniquement ou principalement dans le but de fournir uneinformation :
• concernant un état physiologique ou pathologique
ou
• concernant une anomalie congénitale
ou
• permettant de déterminer la sécurité et la compatibilité avec desreceveurs potentiels
ou
• permettant de contrôler des mesures thérapeutiques.
Les récipients pour échantillons sont considérés comme des dispositifsmédicaux de diagnostic in vitro. On entend par « récipients pour échan-tillons » des dispositifs, qu'ils soient sous vide ou non, spécifiquementdestinés par leur fabricant à recevoir directement l'échantillon provenantdu corps humain et à le conserver en vue d'un examen de diagnostic invitro.
Les produits destinés à des usages généraux en laboratoire ne sont pasdes dispositifs médicaux de diagnostic in vitro à moins que, eu égard àleurs caractéristiques, ils soient spécifiquement destinés par leur fabri-cant à des examens de diagnostic in vitro. »
Exemples de dispositifs médicaux de diagnostic in vitro :
• Dispositif de détection des anticorps
• Test de grossesse
• Bandelette détectant le glucose dans les urines
Exemples de matériel de laboratoire n’étant pas un DMDIV :
• Centrifugeuse
• Balance de précision
4.1.4 Dispositif Médical Actif ou DMADéfinition de la directive 93/42/CEE :
« Tout dispositif médical dépendant pour son fonctionnement d'unesource d'énergie électrique ou de toute autre source d'énergie que cellegénérée directement par le corps humain ou la pesanteur. »
Exemples de dispositifs médicaux actifs* :
• Logiciel médical
• Equipement d’imagerie médical (scanner, IRM…)
• Bistouri électrique
• Endoscope
• Equipement de CEC (Circulation extra-corporelle)
4.1.5 Dispositif Médical Implantable Actif ou DMIADéfinition de la directive 90/385/CEE :

23Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE22
« Tout dispositif médical actif qui est conçu pour être implanté en totalitéou en partie, par une intervention chirurgicale oumédicale, dans le corpshumain ou, par une intervention médicale, dans un orifice naturel et quiest destiné à rester après l'intervention. »
Exemples de dispositifs médicaux implantables actifs :
• Stimulateur cardiaque (pacemaker)
• Neuro-stimulateur
• Pompe à insuline implantable
• Implant cochléaire
4.2 Les directives applicables en fonction du type de dispositifmédical
4.2.1 Les DM, DMDIV, DMA, DMIA
La réglementation des dispositifs médicaux repose sur 3 directives :
• La directive 93/42/CEE pour les dispositifs médicaux et les disposi-tifs médicaux actifs
• La directive 90/385/CEE pour les dispositifs médicaux implantablesactifs
• La directive 98/79/CE pour les dispositifs médicaux de diagnostic invitro
Les directives 90/385/CEE et 98/79/CE sont des directives dites « spéci-fiques » au sens de la nouvelle approche. C'est-à-dire que leurs exi-gences essentielles comprennent les exigences essentielles de ladirective générale 93/42/CEE et qu’il convient de ne tenir compte que desexigences essentielles de la directive spécifique dans le marquage CEdes DMIA et des DMDIV.
Ce guide décrit les procédures demarquage CE des dispositifs médicauxselon la directive 93/42/CEE. Pour les DMIA et les DMDIV, les procéduresapplicables sont différentes et ne sont pas décrites dans ce guide.
En plus de ces directives particulières, d’autres directives européennessont applicables en fonction du dispositif médical considéré. Par exem-ple, sont applicables :
• La directive 87/404/CEE pour les dispositifs médicaux incorporantdes récipients à pression simple (pompes…)
• La directive 2004/108/CE (compatibilité électromagnétique) pour lesdispositifs électro-médicaux
• La directive 2004/22/CE pour les dispositifs médicaux fournissantune mesure
Il est donc toujours indispensable de réaliser une veille réglementaire auniveau européen pour le marquage CE d’un dispositif médical afin des’assurer que toutes les directives applicables en vigueur ont bien étéidentifiées et prises en compte pour l’attestation de la conformité duproduit.
4.2.2 LesDM incorporant unmédicament ou«produits frontières»
Les dispositifs médicaux destinés à l'administration d'un médicament*sont soumis à la directive 93/42/CEE à condition que le médicamentpossède une autorisation demise sur lemarché (AMM) et qu’il soit venduséparément du dispositif médical.
Exemples :
• Seringues rechargeables ou jetables vides
• Matériel de perfusion
Dans le cas où le dispositif médical forme avec lemédicament un produitunique non-réutilisable et que la fonction principale du produit est obte-nue via le médicament, le produit est soumis à la directive 2001/83/CEconcernant lesmédicaments et doit obtenir une AMMpour être commer-cialisé. Il faut, dans ce cas, justifier de la conformité du produit aux exi-gences essentielles de la directive 93/42/CEE dans le dossier dedemande d’AMM.
Exemples :
• Seringues pré-remplies jetables
• Stylos injecteurs d’insuline jetables
• Dispositif d’administration de vaccins jetables
Enfin, dans le cas où le dispositif incorpore un médicament qui l’assistedans sa fonction principale, le dispositif est évalué selon la directive93/42/CEE. Cependant, le médicament incorporé au dispositif médical

25Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE24
doit avoir obtenu préalablement une AMM et l’avis de l’EMEA6 est de-mandé par l’organisme notifié chargé de délivrer lemarquage CE afin des’assurer que l’ajout du médicament est médicalement pertinent.
Exemples :
• Cathéter ou poche à sang enduit d’héparine
• Pansement imprégné de produit bactériostatique
• Implants intravasculaires recouverts de substances actives (médi-caments)
Unmédicament est, selon la définition de la directive 2007/83/CE, « toutesubstance ou composition présentée comme possédant des propriétéscuratives ou préventives à l'égard des maladies humaines. Toute subs-tance ou composition pouvant être administrée à l'homme en vue d'éta-blir un diagnostic médical ou de restaurer, corriger ou modifier desfonctions physiologiques chez l'homme est également considéréecomme médicament ».
Ces substances sont définies comme : « Toute matière quelle qu'en soitl'origine, celle-ci pouvant être :
- humaine, telle que : le sang humain et les produits dérivés du sanghumain,
- animale, telle que : les micro-organismes, animaux entiers, partiesd'organes, sécrétions animales, toxines, substances obtenues parextraction, produits dérivés du sang,
- végétale, telle que : les micro-organismes, plantes, parties de plantes,sécrétions végétales, substances obtenues par extraction,
- chimique, telle que : les éléments, matières chimiques naturelles et lesproduits chimiques de transformation et de synthèse ».
Il est important de faire très attention aux substances incorporées dansun dispositif médical. En effet, un extrait de plante ajouté pour l’odeur oula couleur d’un dispositif peut être considéré par l’autorité compétente*comme un médicament si cette plante est inscrite à la pharmacopéeeuropéenne. Cette plante entraînera alors une reclassification du dispositifmédical en classe de risque supérieur ainsi qu’un allongement dans lecoût et la durée de la procédure de marquage CE.
4.3 La mise à jour des directives 93/42/CEE, 90/385/CEE et98/8/CE
Le 21 septembre 2007 a été publiée au Journal Officiel de L’Union Euro-péenne la directive 2007/47/CEmodifiant la directive 93/42/CEE (relativeaux dispositifs médicaux), la directive 90/385/CEE (relative aux dispositifsmédicaux implantables actifs) et la directive 98/8/CE (relative auxproduits biocides).
4.3.1 Les modifications apportées par la directive 2007/47/CE
4.3.1.1 Les logiciels
La directive 2007/47/CEE met à jour la définition d’un dispositif médicalen y intégrant les logiciels seuls. Cela ne signifie pas que tous les logi-ciels utilisés dans un environnement médical sont des dispositifs médi-caux : seuls les logiciels ayant un objectif thérapeutique, diagnostic ou deprévention d’une maladie sont des dispositifs médicaux.
Les autres parties de la directive ont été adaptées pour intégrer ce nou-veau dispositif médical. On le retrouve notamment dans la classificationcomme un dispositif médical actif et dans les exigences essentielles avecla nécessité de valider les logiciels selon l’état de l’art en prenant encompte l’ensemble du cycle de vie du logiciel.
4.3.1.2 L’évaluation clinique
L’évaluation clinique* d’un dispositif médical est désormais obligatoirequelle que soit la classe du dispositif. Cette évaluation doit permettre nonseulement d’évaluer les effets indésirables liés aux produit et leur pré-valence mais aussi d’évaluer le rapport bénéfice / risques* du produit.L’évaluation clinique peut être menée de trois façons :
• Par une évaluation critique de la littérature scientifique concernantla sécurité, les performances, les caractéristiques et les conditionsd’utilisation du produit lorsqu’une équivalence avec un autre dispo-sitif médical présent sur le marché peut être démontrée et que lesdonnées issues de cette analyse permettent de démontrer laconformité aux exigences essentielles.
3EMEA : Agence Européenne pour l’Evaluation des Médicaments

27Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE26
• Par une analyse des résultats des investigations cliniques*menéessur le dispositif médical.
• Par une combinaison des deux solutions précédentes.
De plus, les fabricants de produits implantables ou de produits de classeIII ont désormais l’obligation de réaliser une étude clinique prospectiverandomisée.
La surveillance des investigations cliniques a été renforcée :
• Les autorités compétentes ont désormais l’obligation de notifier à laCommission Européenne* et aux autres états membres tout refusd’autorisation, demande de modification significative ou demanded’interruption temporaire d’une investigation clinique.
• Les fabricants doivent également notifier à la Commission Euro-péenne et aux autorités compétentes des états membres touteinterruption définitive d’étude clinique pour raisons de sécurité.
4.3.1.3 Relations avec l’organisme notifié
Au niveau de la confidentialité, plusieurs données ne sont plus considé-rées comme confidentielles et peuvent être mises à la disposition dupublic pour assurer sa sécurité. Il s’agit de :
• L’identité de la personne responsable de lamise sur lemarché d’unproduit,
• Des informations de matériovigilance*,
• Des informations sur les certificats délivrés, modifiés, suspendus,annulés ou refusés par les organismes notifiés.
Cette nouvelle disposition implique que les organismes notifiésinforment leur autorité compétente de tous les certificats délivrés,modifiés, suspendus, annulés ou refusés aux fabricants de dispositifsmédicaux.
Une nouvelle disposition contraint également l’organisme notifié àposséder et archiver un exemplaire du dossier technique des dispositifsmédicaux pour lesquels ils ont délivré un certificat (donc à partir de laclasse IIa). Cet exemplaire est à la disposition des autorités compétentesqui peuvent désormais le consulter auprès de l’organisme notifié ou dufabricant.
4.3.1.4 Le management des risques
Plusieurs nouvelles notions devront à présent être prises en compte dansle management des risques :
• Les dispositifs médicaux qui sont également des « machines »doivent inclure dans leur dossier technique la démonstration de laconformité aux exigences essentielles de la directive 98/37/CE(relative aux machines).
• Les dispositifs médicaux qui sont utilisés comme des équipementsde protection individuelle (gants…) doivent inclure dans leur dossiertechnique la démonstration de la conformité aux exigences essen-tielles de la directive 89/686/CEE (relative aux équipements de pro-tection individuelle).
• Les exigences essentielles prennent en compte l’état de connais-sance et le potentiel de l’utilisateur. C'est-à-dire que la sécurité dudispositif devra être démontrée pour tous les utilisateurs potentiels(particulier sans connaissances médicales, personne handicapée,personne âgée…).
• Si des substances dangereuses listées dans l’annexe I de la directive67/548/CEE (relative à la classification, l'emballage et l'étiquetagedes substances dangereuses) sont utilisées, des précautions parti-culières devront être mises en place pour réduire, notamment, lesrisques connus de carcinogénicité*, mutagénicité* ou toxicité pourla reproduction*. L’étiquetage doit mentionner ces substances et ladestruction du produit doit être maîtrisée.
• Les risques liés au retraitement et à la réutilisation d’un dispositifmédical doivent être détaillés dans le dossier technique et mis àdisposition des autorités compétentes dans le cadre de la réflexionde la Commission Européenne sur le retraitement des dispositifsmédicaux. En particulier, l’étiquetage « usage unique » d’un dispo-sitif médical doit être justifié demanière pertinente et doit être uni-forme dans l’ensemble des états membres.
4.3.1.5 La classification
Les règles de classification des dispositifsmédicaux n’ont que peu évolué.

29Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE28
Seuls quelques dispositifs sont concernés par des reclassifications :
• Les dispositifs incorporant des substances dérivées de tissushumains
• Les dispositifs en contact avec le système circulatoire ou le systèmenerveux
• Les dispositifs destinés à désinfecter d’autres dispositifs médicaux.
Parmi les règles de classification, la notion de continuité dans le tempsa été précisée suite à plusieurs problèmes d’interprétation : la continuitédans le temps s’entend si un dispositif reste en place demanière continueou s’il est retiré pour être remplacé par un autre dispositif parfaitementidentique.
De nouvelles procédures internes aux organismes notifiées et aux auto-rités compétentes ont également été prévues pour faire face aux pro-blèmes de classification. En particulier, un fabricant pourra participeraux débats sur la classification de son dispositif en cas de désaccord avecson organisme notifié. Si aucun accord n’est trouvé entre les deux parties,la classification du dispositif sera demandée à la Commission Euro-péenne qui classifiera le produit en fonction des produits existants sur lemarché.
4.3.2 La transposition des directives
Pour que cette directive soit applicable par les fabricants, il est néces-saire que les états membres la transposent dans leur droit national. EnFrance, la directive 2007/47/CE va donc devenir un décret qui modifiera(en particulier) la loi n°94-43 du 18 janvier 1994, la loi n°95-116 du 4février 1995 et le décret n°95.292 du 16mars 1995 transposant la directive93/42/CEE.
Les états membres sont tenus de publier la transposition de la directive2007/47/CE avant le 21 décembre 2008. Les fabricants devront appliquerles nouvelles dispositions (la directive 93/42/CEEmodifiée) à partir du 21mars 2010.
4.3.3 La période transitoireDurant la période transitoire d’application de la directive 2007/47/CE,
c'est-à-dire entre le 21 décembre 2008 et le 21 mars 2010, les fabricantsseront libres de mettre sur le marché des produits marqués CE selonl’ancienne directive 93/42/CEE ou selon la directive 93/42/CEE modifiéepar la directive 2007/47/CE.
Cependant, il est fortement recommandé aux fabricants de réaliser dèsà présent lemarquage CE de leurs dispositifs médicaux selon la directive93/42/CEE modifiée afin de ne pas avoir à recommencer une procédurede marquage CE en 2010. De plus, pour les produits déjà sur le marché,la mise en conformité pouvant être longue (mise à jour du dossier tech-nique, étude clinique en cas de reclassification…), il est égalementrecommandéaux fabricants de la réaliser le plus tôt possible afin de pouvoircontinuer à vendre leurs produits après le 21 mars 2010.

31Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE30
5 - LES ACTEURS DU MARQUAGE CE
5.1 La Commission EuropéenneLa Commission Européenne siège à Bruxelles. Elle représente et défend lesintérêts de l’Union Européenne dans son ensemble et est indépendante desgouvernements nationaux. C’est elle qui élabore les propositions de nou-velles lois européennes qu’elle soumet au Parlement Européen et auConseil. Elle veille au respect des traités européens et de la législation com-munautaire. Elle peut prendre des mesures à l’encontre des contrevenants,et notamment les assigner devant la Cour de Justice Européenne.
La Commission Européenne est l’autorité suprême pour la réglementationdes dispositifs médicaux. Elle est habilitée à trancher un litige entre unfabricant et son autorité compétente. La Commission Européenne évalueles procédures de sauvegarde décidées par les états membres pour lesdispositifs médicaux afin de les étendre aux autres états membres ou, aucontraire, de les rendre invalides.
Les dispositifs médicaux sont gérés par l’unité « Cosmétiques et Dispositifsmédicaux ».
5.2 Les Autorités compétentesL’autorité compétente est l’autorité nationale pour les dispositifs médicaux.Selon les états membres, celle-ci fait partie du Ministère de la Santé ou estun organisme séparé. La mission de l’autorité compétente est d’assurerla sécurité d'emploi, l'efficacité, la qualité et le bon usage des dispositifsmédicaux. Elle assure également la surveillance des effets ou événementsindésirables liés à leur utilisation.
L’autorité compétente n’intervient pas dans le processus de marquage CEqui se réalise sous la responsabilité du fabricant. Son rôle est d’intervenira posteriori pour surveiller le marché. Cette surveillance s’exerce de quatrefaçons :
• Par l’évaluation des incidents et des risques d’incidents qui lui sontsignalés dans le cadre de la matériovigilance
• Par les déclarations demise sur lemarché auxquelles sont soumis lesfabricants

33Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE32
• Par toute action d’évaluation concernant des dispositifs dont il convientde s’assurer de leur conformité aux exigences de santé et de sécurité(tests sur les produits mis sur lemarché, inspections chez le fabricant,évaluation des dossiers techniques de marquage CE…)
• Par des actions d'information auprès des professionnels de santé, dupublic et des industriels pour améliorer le bon usage des dispositifsmédicaux.
L’autorité compétente est également en charge des autorisations d’essaiscliniques impliquant des dispositifs médicaux, menés sur son territoirenational. Elle a la compétence pour prendre les mesures de police sanitaireappropriées sur son territoire, en cas de risque pour la santé publique,à condition d’en tenir informée la Commission Européenne et de suivre laprocédure décrite dans la directive 93/42/CEE.
En France, l’autorité compétente est l’Afssaps*7. La liste des autorités com-pétentes nationales de l’Union Européenne et leurs coordonnées sont dispo-nibles sur le site de l’Union Européenne à l’adresse :
http://ec.europa.eu/enterprise/medical_devices/ca/list_ca.htm
5.3 Les Organismes notifiésUn organisme notifié (ou ON) est un organisme qui est habilité par l’au-torité compétentepourévaluer la conformitéd’undispositifmédical à la directive93/42/CEE afin d’autoriser le fabricant à y apposer le marquage CE lorsquecette autorisation est nécessaire. L’autorité compétente doit vérifier quel’organisme répond aux exigences de compétences demandées par ladirective 93/42/CEE pour le notifier. Celle-ci le contrôle ensuite réguliè-rement et peut lui retirer sa notification si les exigences de compétences nesont plus respectées. En particulier, l’organismenotifié doit évaluer la confor-mité d’un produit de manière :
• Compétente
• Transparente
• Neutre
• Indépendante
• Non discriminatoire
Il existe aumoins un organisme notifié par état membre. Les fabricants peu-vent s’adresser à n’importe quel organisme notifié pour l’évaluation de leurdispositif médical. En aucun cas, ils ne sont obligés de s’adresser à l’orga-nisme notifié de leur état. Cependant, certaines notifications sont partielleset ne concernent qu’un type de dispositif ou qu’une seule procédure demar-quage CE. Il est nécessaire de vérifier que l’organisme notifié choisi possèdebien la notification pour le marquage CE du dispositif concerné et pour laméthode de marquage CE choisie.
Chaque organisme notifié est identifié par un numéro à quatre chiffres.Lorsque l’intervention d’un organisme notifié est nécessaire pour l’appo-sition dumarquage CE, le numéro d’identification de l’organisme notifié estajouté au marquage CE sur le dispositif ainsi que sur les documentsd’accompagnement* (voir Figure 2). Le certificat délivré par un organismenotifié est reconnu dans l’ensemble de l’Union Européenne, quel que soit lepays d’origine de l’organisme notifié.
Logo CE sans interventiond’un organisme notifié
Figure 2 :Exemples de logo CE avec et sans intervention d’un organisme notifié
Logo CE avec interventionde l’organisme notifié 0459
Il n’existe qu’un seul organisme notifié français : le LNE/Gmed dont lenuméro d’identification est le 0459. Le LNE/Gmed est notifié pour tous lesdispositifs médicaux et toutes les procédures de marquage CE. La liste desorganismes notifiés est disponible sur le site internet de l’Union Européenneà l’adresse :
http://ec.europa.eu/enterprise/newapproach/nando/index.cfm?fuseaction=directive.main
(Cliquer sur le nom de la directive ou télécharger le document PDF proposé.)
7Afssaps : Agence Française de Sécurité Sanitaire des Produits de Santé

35Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE34
5.4 Le Fabricant
Est fabricant au sens de la directive 93/42/CEE :
« La personne physique ou morale responsable de la conception, de lafabrication, du conditionnement et de l'étiquetage d'un dispositif en vue de samise sur lemarché en son propre nom, que ces opérations soient effectuéespar cette même personne ou pour son compte par une tierce personne.
Et
La personne physique ou morale qui assemble, conditionne, traite, remet àneuf et/ou étiquette un ou plusieurs produits préfabriqués et/ou leur assignela destination d'un dispositif en vue de sa mise sur le marché en son nompropre. »
Le fabricant est donc la personne ou la société qui met le produit à disposi-tion des utilisateurs sous son nom propre. Un « assembleur » qui assembledes produits marqués CE pour un patient donné (prothésiste dentaire, parexemple) ou un « répartiteur » qui revend des produits sous la marque dufabricant initial ne sont pas des fabricants (voir Figure 3).
Le fabricant est soumis à l’apposition dumarquage CE sur ses dispositifs etil en assume la responsabilité. Suite à la mise sur le marché, le fabricant aun devoir d’information auprès des utilisateurs, de l’autorité compétente etde l’organisme notifié ainsi qu’un devoir de suivi du dispositif, notammentdes incidents ou risques d’incidents liés à celui-ci.
5.5 Le Mandataire dans l’Union EuropéenneEst mandataire* dans l’Union Européenne au sens de la directive93/42/CEE :
« Toute personne physique ou morale établie dans la Communauté qui,après avoir été expressément désignée par le fabricant, agit et peut êtrecontactée par les autorités et les instances dans la Communauté en lieu etplace du fabricant en ce qui concerne les obligations que la présente direc-tive impose à ce dernier ».
Les fabricants qui n’ont pas de siège social dans l’Union Européenne ontl’obligation de désigner un mandataire. Celui-ci, établi dans l’UnionEuropéenne, est le contact des autorités compétentes et de l’organismenotifié choisi par le fabricant. Il gère en particulier l’apposition dumarquage CEet la surveillance du marché. L’autorité compétente fait appel à lui pourobtenir le dossier de marquage CE et pour la gestion des incidents dematériovigilance, notamment dans le cas de rappel de lot.
Le mandataire n’est pas considéré comme un fabricant au sens de ladirective. Les produits sont vendus sous le nom du fabricant établi en dehorsde l’Union Européenne. Le mandataire n’est qu’un contact pour les orga-nismes et les utilisateurs. Il doit être désigné par unmandat écrit le liant aufabricant et détaillant ses missions et responsabilités.
Le mandataire peut être n’importe quel organisme ayant un siège socialdans l’Union Européenne. La structure et l’organisation du mandataire doitlui permettre de répondre à ses obligations réglementaires, notamment enterme de conservation des informations et de matériovigilance. Le manda-taire est audité en même temps que le fabricant lors de la procédure demarquage CE afin de s’assurer que celui-ci respecte ses engagements.
Pour un même produit, un fabricant hors Union Européenne ne peut dési-gner qu’un seul mandataire pour toute l’Europe. L’autorité compétente pource produit sera celle du pays dans lequel le mandataire est installé.
Figure 3 :llustration de la notion de Fabricant au sens de la directive : dans ce schéma,seules les entreprises B, C et D sont des fabricants au sens de la directive etsont soumises au marquage CE

37Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE36
5.6 Les Distributeurs et Agents
Selon les usages des entreprises ou des pays, un « distributeur » peut éga-lement être nommé « importateur » ou « agent ». Le distributeur est celuiqui va commercialiser le produit dans une zone déterminée (une région, unpays, un groupement de pays…). Le distributeur est un client du fabricant,contrairement au répartiteur qui est un prestataire de service :
• Le distributeur achète le produit au fabricant et le revend au clientfinal (hôpital, pharmacie, patient…) en ajoutant une marge sur le prixdu produit.
• Le répartiteur réalise la réception des commandes et l’envoi du produitau client final et fait parfois office de dépôt pour l’entreprise. Il factureses services au fabricant.
Le distributeur ne change pas le nom du produit et ne doit pas réaliser lemarquage CE (s’il change le nom du produit, il devient fabricant et n’est plusdistributeur). Par contre, le fabricant doit s’assurer que le produit possèdeles homologations ou autorisations de mise sur le marché nécessairespour être commercialisé dans la zone visée par le distributeur.
Un contrat doit lier le distributeur au fabricant afin de définir lesmissions etresponsabilités de chacun, notamment pour tous les points réglementairesrendus obligatoires par la directive 93/42/CEE (traçabilité du produit, surveil-lance du marché, notification d’incidents, rappels de lot…). Il appartient aufabricant de vérifier que le distributeur satisfait aux obligations définies parle contrat par des contrôles réguliers.
5.7 Les Sous-traitants et FournisseursLes sous-traitants et fournisseurs sont concernés par le marquage CE carle fabricant, dans le cadre de l’établissement de la conformité du dispositifmédical, doit s’assurer que le produit fourni est conforme à ses spécifica-tions internes, celles-ci permettant la réalisation d’un dispositif médicalrépondant aux exigences de la directive 93/42/CEE. Suivant la classe dudispositif et l’impact du produit ou du service fourni, les exigences pour lesfournisseurs et sous-traitants seront différentes.
Par exemple :
• Le fabricant achète un composant liquide à un fournisseur afin de réa-liser un produit de classe I. Pour chaque lot livré, le fournisseur joint un
certificat d’analyse du composant liquide permettant au fabricant des’assurer de la conformité de ce produit. Ce certificat est suffisant pourassurer la conformité du dispositif médical de classe I ainsi fabriqué.
• Le fabricant fait réaliser un mélange à un laboratoire de chimie afind’intégrer ce mélange dans un dispositif médical de classe IIb. Pourchaque lot livré, le sous-traitant joint le certificat d’analyse du lot livré.Ce certificat ne suffit pas pour prouver la conformité du dispositifmédical de classe IIb. Dans le cas de ce produit, le sous-traitant estconsidéré comme « une chaîne de fabrication » du fabricant. Cettechaîne de fabrication sera soumise aux mêmes exigences que leschaînes de fabrication internes du fabricant. C'est-à-dire que le fabri-cant devra démontrer que son sous-traitant réalise un contrôle desma-tières premières, respecte les procédures de fabrication pour lemélange, que les contrôles sont réalisés par des personnes qualifiées…Pour simplifier cette démarche, il est judicieux, dans ce cas, de choisirdes sous-traitants certifiés (voir chapitre §8).

39Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE38
6 - LA DÉMARCHE DE MARQUAGE CE
L’apposition du marquage CE est la dernière étape d’une procédure régle-mentée basée sur :
• La classification des dispositifs médicaux en fonction de leur utilisa-tion
• L’élaboration d’un dossier technique de marquage CE par le fabricantdestiné à démontrer la conformité du produit aux exigences essen-tielles de l’annexe I de la directive 93/42/CEE
• La référence à un système d’assurance qualité qui assure que la pro-duction de chaque dispositif médical est conforme
• Un rapport bénéfice/risque du dispositif médical favorable
• Des données cliniques et, le cas échéant, des investigations cliniques
• La mise en place d’un système de vigilance (ou matériovigilance)
• Si nécessaire, l’intervention d’une tierce partie : l’organisme notifié.
6.1 Mon produit est-il un dispositif médical ?
Avant toute démarche demarquage CE, il convient de se demander si le dis-positif est un « dispositif médical » au sens de la directive 93/42/CEE (voir lasection §4.1). La frontière entre les produits de santé (médicaments, pro-duits cosmétologiques et dispositifs médicaux) est très mince et une mau-vaise évaluation peut conduire à une requalification par l’autoritécompétente, démarche qui peut s’avérer coûteuse en argent et en image demarque pour le fabricant.
Afin de déterminer si un produit est un dispositif médical, il est nécessairede se référer à l’indication principale du dispositif et à son mode de fonc-tionnement.
Par exemple :
• Un savon de toilette intime est un produit cosmétique
• Un savon de toilette intime contenant des pré-biotiques (non inscrits àla pharmacopée européenne) agissant sur la flore bactérienne afin deprévenir les infections est un dispositif médical
• Un savon de toilette intime contenant un extrait actif de plante (inscrite

41Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE40
à la pharmacopée européenne dans cette indication) pour calmer lesirritations est un médicament.
En cas de doutes, il est recommandé de se faire conseiller par un expertafin de s’affranchir d’une éventuelle requalification.
6.2 Dans quelle classe de risques mon dispositif se situe-t-il ?
Après s’être assuré que le produit est un dispositif médical, il est nécessairede déterminer sa « classe » afin de choisir le mode de marquage CE de cedispositif parmi ceux applicables. Les classes ont été élaborées pour définirle niveau de contrôle nécessaire pour assurer la conformité d’un dispositifmédical. Il existe 4 classes : I, IIa, IIb et III, la classe I concernant les dispo-sitifs pour lesquels le risque est le plus faible et la classe III ceux pour les-quels le risque est le plus élevé. A chaque classe correspond un choixdifférent de procédures d’évaluation de la conformité.
Compte tenu de la très grande diversité des dispositifs médicaux (de l’im-plant au bistouri, du pansement au scanner), des définitions et des règles declassification générales s’appliquant à n’importe quel dispositif ont été éla-borées. Celles-ci se trouvent en annexe IX de la directive 93/42/CEE. Afin depouvoir déterminer la classe du dispositif, il est essentiel que le fabricantait défini au préalable et de façon précise l’usage auquel est destiné le dis-positif médical et son mode d’action.
La classification va dépendre principalement de :
• La durée d’utilisation du dispositif :
- Temporaire : utilisé en continu pendant moins de soixante minutes
- Court terme : utilisé en continu pendant trente jours au maximum
- Long terme : utilisé en continu pendant plus de trente jours
• Le mode de contact avec le corps humain :
- Dispositif non invasif (qui ne pénètre pas à l'intérieur du corps)
- Dispositif invasif par un orifice naturel du corps (ouverture naturelledu corps, surface externe du globe oculaire ou ouverture artificiellepermanente)
- Dispositif invasif de type chirurgical (mis en place à l'aide ou dans lecadre d'un acte chirurgical)
- Dispositif implantable (dispositif implanté en totalité dans le corpsou remplaçant une surface épithéliale ou la surface de l'œil et quireste en place après l'intervention).
• La source d’énergie nécessaire au dispositif (le cas échéant)
• La destination thérapeutique ou diagnostique
Pour réaliser la classification d’un dispositif, il est possible d’utiliser untableau permettant de lister les règles applicables et la classe du dispositifafférente (voir Tableau 2). Dans le cas particulier où plusieurs règles seraientapplicables, la classe la plus élevée sera retenue pour la classification dudispositif. Dans l’exemple du Tableau 2, la classe d’un appareil de radiodia-gnostic émettant des rayons X (radio) est la classe IIb.
8A = Applicable - NA = Non-applicable
1. Dispositifs non invasifs1.1. Règle 1Tous les dispositifs non invasifs font partie de la classe I, sauf si l'unedes règles suivantes est d'application.
A = classe I
1.2. Règle 2Tous les dispositifs non invasifs destinés à conduire ou à stocker dusang, des liquides ou tissus corporels, des liquides ou des gaz en vued'une perfusion, administration ou introduction dans le corps appar-tiennent à la classe IIa :- s'ils peuvent être raccordés à un dispositif médical actif de la classeIIa ou d'une classe supérieure,
- s'ils sont destinés à être utilisés pour le stockage ou la canalisation dusang ou d'autres liquides corporels ou le stockage d'organes, de par-ties d'organes ou tissus corporels.
Dans tous les autres cas, ils appartiennent à la classe I.
NA
1.3. Règle 3Tous les dispositifs non invasifs visant à modifier la composition biolo-gique ou chimique du sang, d'autres liquides corporels ou d'autres li-quides destinés à être perfusés dans le corps appartiennent à la classeIIb, sauf si le traitement consiste en une filtration, une centrifugation ouen échanges de gaz ou de chaleur, auquel cas ils appartiennent à laclasse IIa.
NA
Régle A / NA8 /classe

43Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE42
1.4. Règle 4Tous les dispositifs non invasifs qui entrent en contact avec de la peaulésée :- relèvent de la classe I s'ils sont destinés à être utilisés commebarrièremécanique, pour la compression ou pour l'absorption des exsudats,
- relèvent de la classe IIb s'ils sont destinés à être utilisés principalementpour des plaies comportant une destruction du derme et ne pouvant secicatriser que par deuxième intention,
- appartiennent à la classe IIa dans tous les autres cas, y compris les dis-positifs destinés principalement à agir sur le microenvironnement desplaies.
NA
2. Dispositifs invasifs2.1. Règle 5Tous les dispositifs invasifs en rapport avec les orifices du corps, autresque les dispositifs invasifs de type chirurgical et qui ne sont pas destinésà être raccordés à un dispositif médical actif ou qui sont destinés à êtreraccordés à un dispositif médical actif de classe I :- font partie de la classe I s'ils sont destinés à un usage temporaire,- font partie de la classe IIa s'ils sont destinés à un usage à court terme,sauf s'ils sont utilisés dans la cavité buccale jusqu'au pharynx, dans leconduit auditif externe, jusqu'au tympan ou dans une cavité nasale aux-quels cas ils font partie de la classe I,
- font partie de la classe IIb s'ils sont destinés à un usage à long terme,sauf s'ils sont utilisés dans la cavité buccale jusqu'au pharynx, dans leconduit auditif externe, jusqu'au tympanoudans une cavité nasale et nesont pas susceptibles d'être absorbés par la muqueuse, auxquels casils font partie de la classe IIa.
Tous les dispositifs invasifs en rapport avec les orifices du corps, autresque les dispositifs invasifs de type chirurgical, destinés à être raccordésà un dispositif médical actif de la classe IIa ou d'une classe supérieure,font partie de la classe IIa.
NA
2.2. Règle 6Tous les dispositifs invasifs de type chirurgical destinés à un usage tem-poraire font partie de la classe IIa, sauf :- s'ils sont spécifiquement destinés à contrôler, diagnostiquer, surveillerou corriger une défaillance du cœur ou du système circulatoire centralpar contact direct avec ces parties du corps, auxquels cas ils font partiede la classe III,
- s'il s'agit d'instruments chirurgicaux réutilisables, auquel cas ils fontpartie de la classe I,
- s'ils sont spécifiquement destinés à être utilisés en contact direct avecle système nerveux central, auquel cas ils font partie de la classe III,
- s'ils sont destinés à fournir de l'énergie sous la formede rayonnementsionisants, auquel cas ils font partie de la classe IIb,
- s'ils sont destinés à avoir un effet biologique ou à être absorbés en to-talité ou en grande partie, auxquels cas ils font partie de la classe IIb,
- s'ils sont destinés à administrer des médicaments par un mécanismede libération et que le mode d'administration peut présenter desrisques, auquel cas ils font partie de la classe IIb.
NA
Régle A / NA8 /classe
2.3. Règle 7Tous les dispositifs invasifs de type chirurgical destinés à unusage à courtterme appartiennent à la classe IIa, sauf s'ils sont destinés :- spécifiquement à contrôler, diagnostiquer, surveiller ou corriger unedéfaillance du cœur ou du système circulatoire central par contact di-rect avec ces parties du corps, auxquels cas ils font partie de la classeIII,
ou- spécifiquement à être utilisés en contact direct avec le systèmenerveuxcentral, auquel cas ils font partie de la classe III,
ou- à fournir de l'énergie sous la forme de rayonnements ionisants, auquelcas ils font partie de la classe IIb,
ou- à avoir un effet biologique ou à être absorbés en totalité ou en grandepartie, auxquels cas ils font partie de la classe III,
ou- à subir une transformation chimique dans le corps, sauf s'ils sont placésdans les dents, ou à administrer desmédicaments, auxquels cas ils fontpartie de la classe IIb.
NA
2.4. Règle 8Tous les dispositifs implantables et les dispositifs invasifs à long termedetype chirurgical font partie de la classe IIb sauf s'ils sont destinés :- à être placés dans les dents, auquel cas ils font partie de la classe IIa,- à être utilisés en contact direct avec le cœur, le système circulatoirecentral ou le système nerveux central, auxquels cas ils font partie de laclasse III,
- à avoir un effet biologique ou à être absorbés en totalité ou en grandepartie, auxquels cas ils font partie de la classe III,
- à subir une transformation chimique dans le corps, sauf s'ils sont placésdans les dents, ou à administrer desmédicaments, auxquels cas ils fontpartie de la classe III.
NA
3. Autres règles applicables aux dispositifs actifs3.1. Règle 9Tous les dispositifs actifs thérapeutiques destinés à fournir ou échangerde l'énergie font partie de la classe IIa, sauf si leurs caractéristiques sonttelles qu'ils peuvent fournir de l'énergie au corps humain ou assurer destransferts d'énergie avec celui-ci d'unemanière potentiellement dange-reuse, compte tenu de la nature, de la densité et du site d'application decette énergie, auquel cas ils font partie de la classe IIb.Tous les dispositifs actifs destinés à contrôler et à surveiller les perfor-mances des dispositifs actifs thérapeutiques de la classe IIb ou destinésà agir directement sur les performances de ces dispositifs font partie dela classe IIb.
NA
Régle A / NA8 /classe

45Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE44
3.2. Règle 10Les dispositifs actifs destinés au diagnostic font partie de la classe IIa :- s'ils sont destinés à fournir de l'énergie qui sera absorbée par le corpshumain, à l'exception des dispositifs utilisés pour éclairer le corps dupatient dans le spectre visible,
- s'ils sont destinés à visualiser la distribution de produits radiopharma-ceutiques in vivo,
- s'ils sont destinés à permettre un diagnostic ou un contrôle direct desprocessus physiologiques vitaux, sauf s'ils sont spécifiquement destinésà surveiller les paramètres physiologiques vitaux, lorsquedes variationsde certains de ces paramètres, notamment ceux des fonctions car-diaques ou respiratoires ou de l'activité du système nerveux central,peuvent présenter un danger immédiat pour la vie du patient, auquelcas ils font partie de la classe IIb.
Les dispositifs actifs destinés à émettre des rayonnements ionisants etdestinés au radiodiagnostic et à la radiologie interventionnelle thérapeu-tique, y compris les dispositifs qui commandent ou contrôlent ces dispo-sitifs ou agissent directement sur leurs performances, font partie de laclasse IIb.
A = classe IIb
Règle 11Tous les dispositifs actifs destinés à administrer dans le corps et/ou à ensoustraire desmédicaments, des liquides biologiques ou d'autres subs-tances font partie de la classe IIa, sauf si cette opération est potentielle-ment dangereuse, compte tenu de la nature des substancesadministrées, de la partie du corps concernée et dumode d'administra-tion, auquel cas ils font partie de la classe IIb.
NA
3.3. Règle 12Tous les autres dispositifs actifs font partie de la classe I. NA
4. Règles spéciales4.1. Règle 13Tous les dispositifs incorporant comme partie intégrante une substancequi, si elle est utilisée séparément, peut être considérée comme unmé-dicament au sens de l'article 1er de la directive 2001/83/CE et qui estsusceptible d'agir sur le corps par une action accessoire à celle des dis-positifs font partie de la classe III.Tous les dispositifs incorporant comme partie intégrante une substancedérivée du sang humain font partie de la classe III.
NA
4.2. Règle 14Tous les dispositifs utilisés pour la contraception ou pour prévenir latransmission demaladies sexuellement transmissibles font partie de laclasse IIb, sauf s'il s'agit de dispositifs implantables ou de dispositifs in-vasifs à long terme, auxquels cas ils font partie de la classe III.
NA
Régle A / NA8 /classe
4.3. Règle 15Tous les dispositifs destinés spécifiquement à désinfecter, nettoyer, rin-cer ou, le cas échéant, hydrater des lentilles de contact font partie de laclasse IIb.Tous les dispositifs destinés spécifiquement à désinfecter les dispositifsmédicaux font partie de la classe IIa à moins qu'ils ne soient destinésspécifiquement à désinfecter les dispositifs invasifs auquel cas ils fontpartie de la classe IIb.Cette règle ne s'applique pas aux produits destinés à nettoyer les dis-positifsmédicaux autres que les verres de contact par desmoyens phy-siques.
NA
4.4. Règle 16Les dispositifs destinés spécifiquement à enregistrer les images de ra-diodiagnostic font partie de la classe IIa.
NA
4.5. Règle 17Tous les dispositifs fabriqués à partir de tissus d'origine animale oudedé-rivés rendus non viables entrent dans la classe III, sauf si ces dispositifssont destinés à entrer en contact uniquement avec une peau intacte.
NA
5. Règle 18Par dérogation aux autres règles, les poches à sang figurent dans laclasse IIb.
NA
Régle A / NA8 /classe
Tableau 2 :Exemple de tableau de classification (cas d’un appareil de radiodiagnosticconventionnel)
En cas de litige entre un fabricant et son organisme notifié sur la classifica-tion d’un dispositif médical, c’est l’autorité compétente du pays du fabricant(l’AFSSaPS pour la France) qui détermine la classe du dispositif. Si le fabri-cant n’est pas d’accord avec la classification réalisée par l’autorité compé-tente, un recours est possible auprès de la Commission Européenne quitranchera définitivement le différend.

47Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE46
6.3 Quel mode demarquage CE vais-je choisir ?
Les modalités de marquage CE sont définies dans l’article 11 de la directive93/42/CEE (évaluation de la conformité). Il est nécessaire de souligner l’im-portance de la procédure d’évaluation et donc du choix dumode demarquageCE pour que celui-ci se réalise dans de bonnes conditions.
Pour déterminer la procédure d’évaluation de la conformité de son produit,le fabricant doit prendre en compte :
• la classe du produit
• le type de fabrication (à l’unité, en grande série…)
• l’état d’avancement d’une démarche demanagement de la qualité dansl’entreprise (type ISO 9001)
• le coût et la durée de la procédure.
En effet, l’annexe II (système complet d'assurance de qualité) nécessite lamise en place d’un système complet demanagement de la qualité dans l’en-treprise tandis que les annexes V (assurance de la qualité de la production)et VI (assurance de la qualité des produits) impose lamise en place d’un sys-tème de management de la qualité partiel.
L’annexe II implique, en outre, la vérification du dossier de conception parl’organisme notifié pour les produits de classe III. L’annexe III impose lecontrôle par l’organisme notifié d’un échantillon représentatif de la produc-tion tandis que l’annexe IV implique un contrôle de tous les lots de productionpar l’organisme notifié (ce qui peut se révéler très coûteux à l’usage). L’annexeVII, quant à elle, est nommée « autocertification* » car la conformité est at-testée par le fabricant seul sans intervention d’un organisme notifié. Cepen-dant, la documentation technique du produit doit être maintenue à jour etpeut être demandée à tout moment par l’autorité compétente qui peut éga-lement effectuer des contrôles sur site de manière aléatoire.
Les procédures demarquage CE sont décrites dans l’article 11 de la directive93/42/CEE et se réfèrent aux annexes II à VII de la directive (voir la Figure 4) :
• Annexe II : Déclaration CE de conformité - Système complet d'assurancede la qualité
• Annexe III : Examen CE de type (examen d’un échantillon représentatifdu dispositif)
• Annexe IV : Vérification CE (contrôle de la production pour chaque lot)
• Annexe V : Déclaration CE de conformité - Assurance de la qualité de laproduction
• Annexe VI : Déclaration CE de conformité - Assurance de la qualité desproduits
• Annexe VII : Déclaration CE de conformité (autocertification)
Les fabricants ont théoriquement le choix entre toutes les procédures propo-sées, cependant, dans la pratique, selon la situation de l’entreprise et la na-ture du dispositif médical, une solution stratégiquement plus judicieuses’imposera au fabricant.
Figure 4 :Procédures d’évaluation en fonction de la classe du dispositif médical
49Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE48
6.4 Quel organisme notifié vais-je choisir ?
Le choix de l’organisme notifié va principalement dépendre de trois critèresdont l’importance relative sera déterminée par le fabricant. Ces trois critèressont :
• La langue d’évaluation
• Le coût de l’évaluation
• Le délai de réalisation de l’évaluation.
Ces critères sont extrêmement variables d’un organisme notifié à un autre,c’est pourquoi il est préférable de demander un devis avec délai à l’ensembledes organismes ayant la notification pour le produit et la procédure d’évalua-tion, afin de choisir la meilleure proposition en fonction des contraintes del’entreprise.
6.5 Planification d’une démarche de marquage CE
La durée d’une démarche de marquage CE va dépendre de l’organismenotifié choisi, de la classe du dispositif et de la méthode de marquage CEsélectionnée. Il est nécessaire de planifier cette démarche en amont afin delimiter la durée de celle-ci et de fluidifier la démarche qui peut s’avérer trèslongue (de 3 mois à plusieurs années).
La Figure 5 est donnée à titre d’exemple. Les durées mentionnées sont desdurées moyennes qui évoluent nécessairement en fonction :
• Des ressources liées au marquage CE
- Affectation de personnel qualifié pour cette démarche
- Intervention d’une ressource extérieure (conseiller, expert…)
- Disponibilité de l’organisme notifié
- …
• Des étapes intermédiaires à réaliser dont les durées peuvent être trèsvariables
- Par exemple, compter 3 mois pour les essais de biocompatibilité*simples contre 1 à 2 ans pour les essais d’implantation sur animaux
- La durée des essais de fatigue* et des essais cliniques dépendent dela durée de vie ou d’utilisation du dispositif médical.
- …
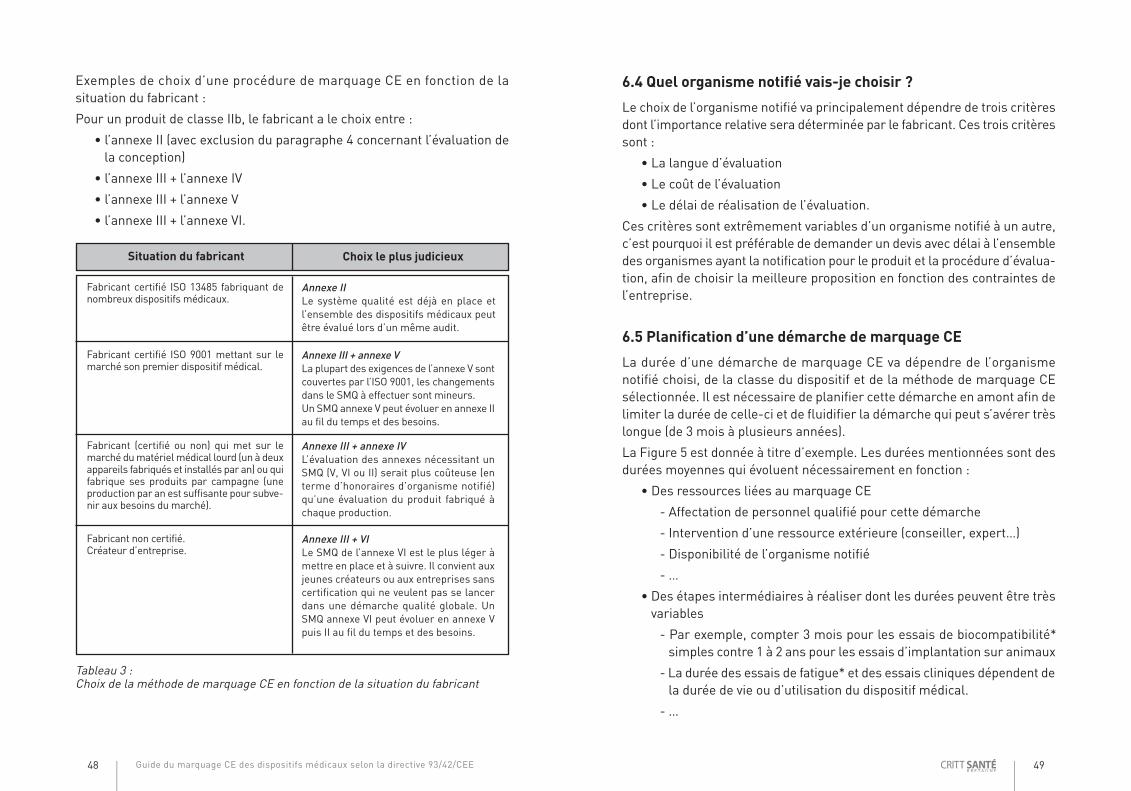
Exemples de choix d’une procédure de marquage CE en fonction de lasituation du fabricant :
Pour un produit de classe IIb, le fabricant a le choix entre :
• l’annexe II (avec exclusion du paragraphe 4 concernant l’évaluation dela conception)
• l’annexe III + l’annexe IV
• l’annexe III + l’annexe V
• l’annexe III + l’annexe VI.
Fabricant certifié ISO 13485 fabriquant denombreux dispositifs médicaux.
Annexe IILe système qualité est déjà en place etl’ensemble des dispositifs médicaux peutêtre évalué lors d’un même audit.
Fabricant certifié ISO 9001 mettant sur lemarché son premier dispositif médical.
Annexe III + annexe VLa plupart des exigences de l’annexe V sontcouvertes par l’ISO 9001, les changementsdans le SMQ à effectuer sont mineurs.Un SMQ annexe V peut évoluer en annexe IIau fil du temps et des besoins.
Fabricant (certifié ou non) qui met sur lemarché dumatérielmédical lourd (un à deuxappareils fabriqués et installés par an) ou quifabrique ses produits par campagne (uneproduction par an est suffisante pour subve-nir aux besoins dumarché).
Annexe III + annexe IVL’évaluation des annexes nécessitant unSMQ (V, VI ou II) serait plus coûteuse (enterme d’honoraires d’organisme notifié)qu’une évaluation du produit fabriqué àchaque production.
Fabricant non certifié.Créateur d’entreprise.
Annexe III + VILe SMQ de l’annexe VI est le plus léger àmettre en place et à suivre. Il convient auxjeunes créateurs ou aux entreprises sanscertification qui ne veulent pas se lancerdans une démarche qualité globale. UnSMQ annexe VI peut évoluer en annexe Vpuis II au fil du temps et des besoins.
Situation du fabricant Choix le plus judicieux
Tableau 3 :Choix de la méthode de marquage CE en fonction de la situation du fabricant

51Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE50
De plus, certaines règles et délais doivent être impérativement prises encompte pour planifier la démarche de marquage CE :
• Il est nécessaire d’attendre au moins 3 mois entre la mise en placeeffective du système qualité et l’audit de l’organisme notifié afin quecelui-ci puisse l’auditer dans de bonnes conditions.
• Si le dossier technique doit être revu par l’organisme notifié (classe IIbet III), il faut compter environ trois mois de délai pour cette vérificationet l’audit du système de management de la qualité ne peut se fairequ’une fois cette vérification achevée.
7 - LE DOSSIER TECHNIQUE DE MARQUAGE CE
Le dossier technique de marquage CE est décrit dans l’annexe VII de ladirective 93/42/CEE ainsi qu’au point 3 de l’annexe III. Sa rédaction est obli-gatoire, quelle que soit la classe du dispositif. Dans le cas d’unmarquage CEréalisé via l’annexe II, le dossier technique n’est pas obligatoirement forma-lisé en tant que dossier unique mais les informations nécessaires au mar-quage CE du produit peuvent être rassemblées via le dossier deconception et l’ensemble des enregistrements du système de manage-ment de la qualité.
Le dossier technique doit être mis à la disposition de toute autorité compé-tente qui en ferait la demande, dès la mise sur le marché du produit. Il peutégalement être demandé par la Commission Européenne ou les douanes,lors du passage de frontières (hors Union Européenne). Lors d’une demande,le délai d’envoi du dossier technique est de l’ordre d’une semaine. C’est laraison pour laquelle il doit être réalisé avant la mise sur le marché du dis-positif médical et mis à jour régulièrement.
Le dossier technique de marquage CE, ainsi que tous les éléments dusystème qualité autorisant le marquage CE, doivent être conservés pendantla durée de vie du dispositif et au moins 5 ans après la dernière mise sur lemarché du produit ou 15 ans dans le cas particulier des dispositifs implan-tables.
Le dossier technique demarquage CE doit êtremis à jour à chaque change-ment dans le procédé de fabrication ou dans la conception du dispositif. Dansle cadre d’un dispositif médical de classe IIb ou III, il est nécessaire d’avertirl’organisme notifié qui a émis le certificat demarquage CE de lamodificationafin qu’il évalue à nouveau le dossier technique et délivre un nouveau certi-ficat, si nécessaire. Le dossier technique de marquage CE doit égalementêtre implémenté avec les données issues dumarché, notamment, en ce quiconcerne les données cliniques et l’analyse des risques*. Il doit être entiè-rement revu au moins tout les cinq ans, durée de validité du marquage CE.
7.1 Les exigences essentielles de la directive 93/42/CEE
Le dossier technique de marquage CE permet à un fabricant de démontrerque son dispositif médical répond à l’ensemble des exigences essentielles
Figure 5 :La planification d’un projet de marquage CE

53Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE52
énoncées dans l’annexe I de la directive 93/42/CEE. Ces exigences essen-tielles permettent d’assurer la sécurité et les performances de l’ensembledes dispositifs médicaux mis sur le marché.
Les exigences essentielles sont classées en exigences générales et enexigences relatives à la conception et à la construction. Ces dernièresconcernent :
• Les propriétés chimiques, physiques et biologiques
• L’infection et contamination microbienne
• Les propriétés relatives à la fabrication et à l'environnement
• Les dispositifs ayant une fonction de mesurage
• La protection contre les rayonnements
• La protection contre les risques électriques
• Les informations fournies par le fabricant.
Les normes harmonisées présentent des moyens pour répondre à cesexigences (voir le Tableau 4). Il est essentiel pour le fabricant de prendre encompte dès la conception de son dispositif médical l’ensemble des exigencesessentielles afin de s’assurer que le produit fabriqué pourra être marquéCE.
2. Les solutions choisies par le fabricant dans la conception et la construc-tion des dispositifs doivent se tenir aux principes d'intégration de la sécu-rité en tenant compte de l'état de la technique généralement reconnu.Pour retenir les solutions les mieux appropriées, le fabricant doit appli-quer les principes suivants dans l'ordre indiqué :- éliminer ou réduire autant que possible les risques (sécurité inhérenteà la conception et à la fabrication),
- le cas échéant, prendre lesmesures de protection appropriées, y com-pris des dispositifs d'alarme au besoin, pour les risques qui ne peuventêtre éliminés,
- informer les utilisateurs des risques résiduels dus à l'insuffisance desmesures de protection adoptées.
EN ISO 14971
6 bis. La démonstration de la conformité aux exigences essentielles doitinclure une évaluation clinique conformément à l'annexe X. EN ISO 14155
8.4. Les dispositifs qui sont délivrés en état stérile doivent avoir été fabri-qués et stérilisés selon uneméthode appropriée et validée.
EN 556EN ISO 11135EN ISO 11137EN ISO 11138EN ISO 11140EN ISO 11607EN ISO 11737EN 13824
EN ISO 14937EN 15424
EN ISO 17665
12.6. Protection contre les risques électriquesLes dispositifs doivent être conçus et fabriqués de façon à éviter, danstoute lamesure du possible, les risques de chocs électriques accidentelsdans des conditions normales d'utilisation et en condition de premier dé-faut, lorsque les dispositifs sont correctement installés.
EN 60601
13.1. Chaque dispositif doit être accompagné des informations néces-saires pour pouvoir être utilisé correctement et en toute sécurité, en te-nant compte de la formation et des connaissances des utilisateurspotentiels et pour permettre d'identifier le fabricant.Ces informations sont constituées des indications figurant dans la noticed'instruction.Dans lamesure où cela est possible et approprié, les informations néces-saires pour utiliser le dispositif en toute sécurité doivent figurer sur ledispositifmêmeet/ou sur l'emballage de chaqueunité ou, le cas échéant,sur l'emballage commercial. S'il n'est pas possible d'emballer séparé-ment chaque unité, les informations doivent figurer sur une notice ac-compagnant un ou plusieurs dispositifs.L'emballage de chaque dispositif doit contenir une notice d'instruction.Une exception est faite pour les dispositifs des classes I et IIa, s'ils peu-vent être utilisés en toute sécurité sans l'aide de telles instructions.
EN 980EN 1041
EN ISO 17664
Exigence Essentielle Normeharmonisée
Tableau 4 :Exemples d’exigences
essentielles et de normesharmonisées y répondant

55Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE54
7.2 Les parties principales du Dossier Techniquede Marquage CE
Le dossier technique doit démontrer la conformité du dispositif médical auxexigences essentielles, c'est-à-dire qu’il doit démontrer son innocuité et sesperformances. Si le contenu d’un dossier technique varie en fonction du typede dispositif, les parties décrites dans ce chapitre restent indispensables.Elles seront plus ou moins détaillées, le cas échéant.
7.2.1 Identification du fabricant et de son représentant
Cette partie comprend une description juridique de la société (coordon-nées, n° SIRET…) ainsi qu’une description « marketing » permettant demettre en avant les différentes activités de la société et les produitsfabriqués. C’est dans ce chapitre qu’est désigné le responsable de lamise sur le marché ainsi que le mandataire dans l’Union Européenne, sicelui-ci est nécessaire pour la mise sur le marché du dispositif médicalconcerné.
Généralement, ce chapitre introductif comprend également une déclara-tion d’engagement du responsable de la mise sur le marché sur :
• La véracité des informations présentes dans le dossier technique
• Le respect des exigences essentielles
• La mise à jour régulière du dossier technique
• L’information de l’organisme notifié en cas de changement interve-nant sur le produit ou le système qualité
• L’exclusivité de la demande de certification auprès de l’organismenotifié.
7.2.2 Identification du dispositif médical et de ses variantes
Cette partie comprend une description technique du dispositif, de sescomposants (pièces détachées, ingrédients, matières…) y compris duconditionnement. Les différentes variantes (formes, tailles, couleurs,conditionnements…) dumême dispositif doivent être décrites et les réfé-rences explicitées. Il est fortement recommandé d’illustrer cette partieavec des schémas techniques et / ou des photos du dispositif et des élé-ments qui le composent ce qui permet demieux percevoir le dispositif et
ses variantes. Les fiches de spécification du produit fini sont générale-ment mentionnées dans cette partie et jointes en annexe.
L’usage revendiqué du dispositif, c'est-à-dire son utilisation, doit êtredécrit. En particulier, seront mentionnées :
• Ses revendications de performance
• Ses indications
• Ses contre-indications
• La posologie
• Le mode d’utilisation.
Dans ce chapitre sont également mentionnées la classification dudispositif et les règles utilisées pour sa détermination. Le tableau declassification présenté au paragraphe §6.2 peut être incorporé. Une dé-claration doit être ajoutée mentionnant si le produit contient ou pas :
• Un médicament
• Un produit d’origine animale
• Un produit dérivé du sang humain.
7.2.3 Dossier de recherche et développement
Le dossier de recherche et développement doit être inclus ou référencédans le dossier technique. Selon le niveau de risque du produit, celui-cisera plus oumoins détaillé : le dossier le plus complet sera réalisé selonl’annexe II pour les produits de classe III.
Ce dossier doit comporter au minimum :
• Le cahier des charges initial du dispositif et les modificationsapportées en cours de développement
• Le planning daté de la recherche et développement
• Un état de l’art des produits existants ayant la même indication
• Les résultats des essais réalisés durant la recherche et le dévelop-pement.

57Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE56
7.2.4 Dossier de fabrication
Le dossier de fabrication doit faire partie intégrante du dossier de mar-quage CEmême si l’entreprise possède un système qualité certifié. Il estindispensable d’y faire apparaître l’ensemble des intervenants dans lafabrication du produit fini (de l’achat des matières premières à l’expédi-tion chez le client) ainsi que les opérations qu’ils réalisent et les certifi-cations qu’ils ont obtenues. Ceci permet, entre autre, à l’organismenotifié d’évaluer les sous-traitants à auditer en sus du fabricant pour dé-livrer le certificat de marquage CE.
Un diagramme des flux* de fabrication couramment appelé « flow-chart*de fabrication » doit également figurer dans cette partie avec lescontrôles à chaque niveau de fabrication. Les protocoles et les critèresd’acceptation de chaque contrôle doivent être succinctement décrits. Desexemples de fiches de contrôle ainsi que les certificats matières pre-mières (s’ils existent) peuvent être joints en annexe. Si des tâches sonteffectuées dans des conditions d’environnement particulières (salleblanche, flux laminaire, laboratoire certifié), elles doivent apparaître dansle flow-chart de fabrication.
Enfin, cette partie contient la description de la chaîne de traçabilité.Celle-ci peut être intégrée dans le flow-chart de fabrication ou décritedans un document séparé. La description de la chaîne de fabrication doitidentifier :
• Les documents conservés à chaque étape
• Les informations présentes dans le dossier de lot
• La façon dont est construit le numéro de lot
• Les éléments permettant de remonter la chaîne de traçabilité et leniveau pouvant être atteint.
Pour la majorité des dispositifs médicaux, le fabricant doit avoir la maî-trise complète de la traçabilité depuis lamatière première jusqu’au clientfinal.
7.2.5 Documents d’accompagnement
Les documents d’accompagnement comprennent tout ce qui accom-pagne le dispositif, c'est-à-dire la notice d’utilisation, le mode d’emploi,
le conditionnement, l’étiquetage, la documentationmarkéting, les fichesproduits… L’ensemble de ces documents doit être joint au dossier tech-nique car ils sont réglementés et doivent pouvoir être vérifiés facilementpar l’organisme notifié et les autorités compétentes. Ces documents doi-vent être rédigés dans la langue nationale des pays dans lesquels seradistribué le dispositif médical. Toutefois, seule la version dans la languedu dossier technique est nécessaire pour celui-ci.
7.2.6 Liste des normes et référentiels revendiqués
Cette partie doit lister les normes harmonisées et les textes réglemen-taires auxquels il a été fait référence dans la conception et la fabricationdu dispositif et dans l’élaboration du dossier technique. On y retrouve no-tamment :
• La directive 93/42/CEE
• Les textes applicables concernant la matériovigilance
• Les normes harmonisées de la directive 93/42/CEE.
Seules les normes citées dans cette partie (et donc appliquées) pourrontservir dans la partie « 7.2.12 Revue de conformité aux exigences essen-tielles » pour démontrer la conformité du dispositif. Le fabricant doit êtreen possession des versions en vigueur des normes auxquelles il faitréférence (ces normes sont payantes, elles peuvent être achetées auprèsdes organismes de normalisation tels que l’AFNOR ou l’ISO).
Si certaines parties des documents mentionnés ne sont pas appliquéespar l’entreprise, il convient de le justifier demanière adéquate et d’expli-citer les méthodes mises en œuvre pour remplacer les parties nonappliquées.
7.2.7 Analyse de la biocompatibilité
L’analyse de la biocompatibilité est obligatoire pour tout dispositif encontact direct avec le corps humain, même s’il est destiné à n’être encontact qu’avec de la peau saine. Les essais de biocompatibilité à réaliservont dépendre :
• Du type de contact avec le corps humain
• De la durée de contact avec le corps humain

59Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE58
• De la biocompatibilité des matières premières
• Du procédé de fabrication.
La norme harmonisée concernant la biocompatibilité est la norme ISO10993 divisée en plusieurs parties (dix-huit à ce jour). Dans la premièrepartie (ISO 10993-1), des tableaux d’aide à la décision permettent dedéterminer les essais de biocompatibilité nécessaires pour un dispositif.Ces essais sont les essais minimum requis pour le marquage CE d’undispositif médical. Si le fabricant ne souhaite pas réaliser certains de cesessais, il doit le justifier de manière scientifique dans le dossier de mar-quage CE.
Les essais de biocompatibilité sont à réaliser sur le produit fini qui doitavoir subi l’ensemble des étapes du procédé de fabrication, de la récep-tion des matières premières à l’emballage final et à la stérilisation, si leproduit est destiné à être vendu stérile. Il est nécessaire que les échan-tillons ou prototypes testés soient parfaitement similaires au dispositifmédical qui va être commercialisé. Si les différentes variantes d’unmême dispositif médical nécessitent des étapes de fabrication différentesou un temps de contact avec le corps humain supérieur, les essais debiocompatibilité devront être réalisés sur la variante du produit la plusrisquée.
Pour les produits de classe IIb et III, ces essais doivent être réalisés par :
• un laboratoire accrédité pour ces essais (accréditation Cofrac enFrance, par exemple)
• un laboratoire reconnu par l’organisme notifié
• ou l’organisme notifié lui-même
car selon la directive, les essais doivent être réalisés par l’organismenotifié lui-même qui atteste donc des résultats présentés.
Les rapports d’essais sont généralement joints au dossier de marquageCE en annexe tandis que le dossier lui-même comprend :
• Le résumé du protocole
• Les critères d’acceptation
• Les résultats obtenus
• La conclusion sur la biocompatibilité du produit.
Il est fortement recommandé de demander l’ensemble des rapportsd’essais en Anglais afin de faciliter l’accès auxmarchés étrangers (Etats-Unis, Canada, Australie, Japon…) après l’obtention du marquage CE.
Pourront être inclus dans cette partie d’éventuels essais cliniques suranimaux si de tels essais ont été réalisés. Ils ne sont pas obligatoirespour l’obtention du marquage CE car l’Union Européenne tend à limiterles essais cliniques de fonctionnalité sur les animaux : si le produit estbiocompatible, sa performance ne sera évaluée que chez l’humain.
7.2.8 Etudes et essais techniques
Cette partie du dossier technique regroupe l’ensemble des études etessais physico-chimiques nécessaires pour la validation du produit. Lesnormes verticales, spécifiques à un produit ou à une catégorie deproduits, permettent de définir les essais à réaliser pour un dispositifmédical donné.
De la même façon que pour les tests de biocompatibilité, les essaisdoivent être réalisés par des laboratoires accrédités pour les produits declasse IIb et III, les rapports d’essais doivent être mis en annexe et unrésumé des essais est présenté dans le dossier technique.
Exemples d’études et essais présentés dans cette partie :
• Analyse des éléments finis*
• Essais de compatibilité électromagnétique
• Essais de fatigue
• Essais de vieillissement
• Essais de validation du conditionnement
• Essais de résistance mécanique (traction, compression, torsion,choc…)
• Analyse des métaux lourds
• Essais de fonctionnement simulé
• Essais de compatibilité avec d’autres dispositifs médicaux
• Analyse des résidus de fabrication (recherche de trace de solvants,d’huile…).

61Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE60
7.2.9 Revue des données cliniques
L’objectif de la revue des données cliniques* est d’évaluer la sécurité etles performances du dispositif en utilisation clinique et d’identifier leséventuels effets secondaires. Les données cliniques peuvent être issues :
• d’essais cliniques réalisés sur le dispositif médical concerné par lemarquage CE
• d’essais cliniques réalisés sur un dispositif dont l’équivalence avecle dispositif médical à marquer CE peut être démontrée.
En pratique, une revue exhaustive de la littérature scientifique concer-nant la pathologie pour laquelle le dispositif est indiqué ainsi que les dis-positifs médicaux équivalents et incluant une analyse critique est réaliséedès la conception du dispositif médical afin de prendre en compte « l’étatde l’art ». Une fois le produit prêt à être mis sur le marché, le fabricantdoit déterminer si cette revue de la littérature est suffisante pour carac-tériser l’innocuité et les performances de son dispositif médical ou si uneétude clinique (prospective, randomisée) est nécessaire.
Dans le cas d’un dispositif médical innovant, de par son matériau, sondesign ou son utilisation, une étude clinique sera toujours nécessaire carl’équivalence avec les produits déjà disponibles sur le marché et pourlesquels des données cliniques existent ne pourra pas être démontrée.
Si une étude clinique est nécessaire pour apposer lemarquage CE sur undispositif, le fabricant doit suivre la procédurementionnée à l’annexe VIIIpourmettre à disposition son produit auprès des professionnels de santédans le cadre de la réalisation de cette étude. De plus, il doit engager, enparallèle, la procédure de demande d’autorisation d’une recherche bio-médicale, procédure qui peut durer jusqu’à un an en France.
7.2.10 Analyse des risques
L’analyse des risques est une partie très importante du dossier tech-nique. Elle s’insère dans la gestion des risques globale de l’entreprise,conformément à la norme ISO 14971. L’objectif de l’analyse des risquespour un dispositif médical est de déterminer les dangers liés à ce dis-positif et les actions à mettre en place pour réduire au maximum cesdangers (ou risques).
Une analyse des risques se déroule en 5 temps :
1. Identification du dispositif médical, de son utilisation, de ses mésu-sages raisonnablement prévisibles (posologie non respectée, utilisa-tion avec d’autres produits de santé…) et de ces caractéristiques,susceptibles d’affecter sa sécurité (état stérile, formulation, montagenécessaire…).
2. Identification des moyens mis en œuvre pour l’analyse des risques(moyens humains, matériels…).
3. Identification de l’ensemble des phénomènes dangereux liés au dis-positif et des conditions dans lesquelles ils peuvent survenir. Lanorme ISO 14971 présente des méthodes pour faciliter la recherchedes phénomènes dangereux : liste de questions, classification desphénomènes dangereux… Il est nécessaire d’avoir réalisé la revue desdonnées cliniques avant l’analyse des risques car elle permet d’iden-tifier l’ensemble des effets indésirables connus liés à un dispositifmédical.
4. Cotation des risques. Les risques sont cotés en fonction de la fré-quence d’apparition du danger et de la gravité du dommage causé àl’homme. Ces deux critères de cotation peuvent être complétés par lecritère de « probabilité de non détection du danger ». Le croisement deces informations permet de définir si un risque est directementacceptable ou s’il doit être réduit. L’acceptabilité d’un risque s’évaluepar rapport au bénéfice apporté au patient : unmême risque peut êtreacceptable pour un implant vasculaire qui sauve la vie du patient alorsqu’il peut ne pas l’être pour un pansement de plaie simple.
5. Conclusion sur la balance bénéfices/risques. Pour qu’un dispositif mé-dical puisse être marqué CE, il doit apporter plus de bénéfices que derisques pour le patient, autrement dit, sa balance bénéfices/risquesdoit être positive : les risques doivent être acceptables au regard desbénéfices apportés. Cette évaluation ne doit pas être subjective maisau contraire documentée.
Les moyens pour réduire les risques sont divers et peuvent aller d’unesimple mise en garde de l’utilisateur jusqu’à un changement de concep-tion du dispositif médical. Chaque action mise en œuvre doit être revuepour en évaluer son efficacité. L’analyse des risques n’est jamais figée,elle doit être mise à jour régulièrement avec les informations provenant

63Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE62
dumarché : incidents liés à l’utilisation du dispositif ou d’un dispositif si-milaire, réclamations et remarques des utilisateurs et données cli-niques notamment.
L’analyse des risques peut conduire à réaliser de nouveaux essais debiocompatibilité ou physico-chimiques ou même déclencher une étudeclinique si un risque ne peut pas être coté ou demeure inacceptable.L’analyse des risques permet, entre autre, de confirmer que l’ensembledes éléments fournis dans le dossier technique sont suffisants pourdémontrer l’innocuité et les performances du dispositif.
7.2.11 Suivi après mise sur le marché
De par l’apposition du marquage CE, le fabricant s’engage à surveiller ledispositif médical durant toute sa durée de mise sur le marché. Cettesurveillance comprend la matériovigilance et la « post-market surveil-lance » ou PMS*.
La matériovigilance consiste à recueillir, analyser et déclarer (si besoin)l’ensemble des incidents ou risques d’incidents survenus lors de l’utili-sation du dispositif médical (voir chapitre 10.1). La PMS consiste à re-cueillir, à analyser et à traiter les informations concernant les produitssimilaires présents sur le marché et les données cliniques (voir chapitre10.2). Dans cette partie du dossier technique, le fabricant doit détailler lesdispositions mises en œuvre pour répondre à ces deux exigences, c'est-à-dire résumer et faire référence aux procédures du système de mana-gement de la qualité mises en place pour la matériovigilance et la PMSou les inclure directement dans le dossier technique si aucun Système deManagement de la Qualité (SMQ) n’est mis en place dans l’entreprise.
7.2.12 Revue de conformité aux exigences essentielles
C’est la dernière partie du dossier technique de marquage CE et la plusimportante. Elle permet de déterminer si l’ensemble des informationsfournies dans le dossier technique permettent de répondre aux exigencesessentielles de la directive 93/42/CEE. Elle se présente généralementsous la forme d’un tableau similaire au Tableau 5.
En conclusion de la revue de conformité aux exigences essentielles, lefabricant réalisera une déclaration de conformité de son produit aux exi-gences de la directive 93/42/CEE. Cette déclaration sera envoyée à l’au-torité compétente au moment de la mise sur le marché pour un produitde classe I et après réception des certificats délivrés par l’organismenotifié pour les produits des classes supérieures. Un deuxième originalde cette déclaration CE de conformité doit être conservé par le fabricantet joint au dossier technique. Il atteste de la conformité du produit auprèsdes utilisateurs, dans les appels d’offres des hôpitaux publics et auprèsdes distributeurs. Il est également demandé, pour les dispositifs declasse I et IIa lors de la demande d’autorisation de mise sur le marchédans les pays hors Union Européenne.
Une fois la déclaration duproduit effectuée auprès de l’autorité compétente,le fabricant apposera le symbole CE sur le dispositif médical et ses docu-ments d’accompagnement et pourra lemettre sur lemarché de l’ensembledes états membres de l’Union Européenne. Le symbole CE apposé doit :
• Etre conforme au symbole de l’annexe XII de la directive 93/42/CEE
• Avoir une hauteur de 5mmminimum
• Avoir une taille en rapport avec la taille du dispositifmédicalmarquéCE.
Dans cettecolonne figurechacunedes exigencesessentiellesde l’annexe 1de la directive93/42/CEE.
A = ApplicableNA = NonApplicable.Cette colonnemet en évidencel’applicabilitéd’une exigenceessentielle pourle dispositif mé-dical concerné.En cas de non-application, unejustification doitêtre fournie.
Cette colonneliste les normesharmoniséesutilisées pourrépondre àl’exigence.L’utilisation d’unenorme harmoni-sée donne uneprésomptionde conformité àl’exigenceessentielle.
Cette colonnefait référenceaux rapportsd’essaisdémontrant quele produit estconformeà la normeharmonisée.
Cette colonnedétaille lesmoyensmisenœuvre pourrépondre àl’exigencelorsque lesnormesharmoniséesn’ont pas étéappliquéesou qu’ellesn’existent paspour cedispositif.
ExigenceEssentielle(Annexe I dela Directive93/42/CEE)
A / NANormes
harmoniséesrevendiquées
Rapportsd’évaluation
Preuve deconformité
Tableau 5 : Tableau d’évaluation de la conformité aux exigences essentielles

65Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE64
7.3 La constitution du dossier technique de marquage CE
La chronologie de constitution d’un dossier de marquage CE est présentéedans la Figure 6.
8 - LA CERTIFICATION D’ENTREPRISE
Pour lemarquage CE des dispositifs médicaux autres que ceux de la classe I,la mise en place d’un système de management de la qualité dans l’entre-prise, total ou partiel, ainsi que sa certification par un organisme notifié, estobligatoire (excepté pour l’annexe IV). Il peut être mis en place à partir desdifférentes annexes de la directive 93/42/CEE (annexes II, V ou VI) ou à partirde la norme ISO 13485 (norme harmonisée pour cette exigence).
La norme ISO 13485 est construite sur lemodèle de la norme ISO 9001 : leurtexte est similaire, seules les exigences réglementaires des dispositifs mé-dicaux ont été intégrées dans la norme ISO 13485. L’avantage de mettre enplace et de faire certifier son entreprise selon la norme ISO 13485, en plusde l’intérêt commercial qu’elle représente, est que cette norme est reconnueà l’international pour l’enregistrement des dispositifs médicaux hors del’Union Européenne.
L’objectif principal du système d’assurance de la qualité (SMQ) est degarantir l’application de procédures validées pour la conception, la productionet le suivi du dispositif médical afin d’assurer que chaque dispositif médicalsortant de l’entreprise est conforme, quelle que soit sa date de fabrication.
Cette partie présente le système de management de la qualité à mettre enplace en fonction de l’annexe choisie : un SMQ complet est nécessaire pourl’annexe II tandis que les annexes V et VI impose la mise en place d’un SMQpartiel. Les autres annexes, traitant de l’évaluation CE du dispositif médical,seront détaillées dans le chapitre suivant.
8.1 Annexe II de la directive 93/42/CEE ou l’ISO 13485
L’annexe II impose lamise en place et le maintien d’un système demanage-ment de la qualité total, c'est-à-dire la mise en place de l’ensemble des pa-ragraphes de la norme ISO 13485. Ces paragraphes et leurs exigences sontdétaillés ci-après.
Dans le cas particulier des dispositifs de classes IIa et IIb, la partie 7.3 de lanorme concernant la conception et le développement peut être exclue dusystème de management de la qualité.
Définition du dispositif médicalet de son utilisation, a priori
Réalisation de la revue desdonnées cliniques
Réalisation de l’analysedes risques
Réalisation des essais
Rédaction du dossier technique
Etat de l’artIdentification des
complications connues
Identification des testset essais à réaliser
Identification desperformances du dispositif
Mise à jour de l’analysedes risques et rédaction du
dossier technique demarquage CE
VALIDATION
CONCEPTION
Figure 6 : Constitution d’un dossier de marquage CE

67Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE66
8.1.1 Système de Management de la Qualité (§49)
Le fabricant doit organiser son entreprise en « processus » cohérentsqui sont surveillés séparément par le Système de Management de laQualité ou SMQ. Chaque processus est décrit avec :
• Ses données d’entrée
• Ses données de sortie
• Son responsable
• Ses indicateurs
• Ses ressources
• Ses procédures, instructions de travail, formulaires et enregistre-ments.
Les exigences selon les processus sont plus ou moins importantes enfonction de l’impact du processus sur le produit fini. Par exemple, les exi-gences concernant les achats, la production et les ressources humainessont plus nombreuses que les exigences concernant la commercialisa-tion. Si une partie d’un processus est externalisée (stérilisation réaliséechez un sous-traitant, par exemple), le fabricant doit pouvoir en assurerla conformité à la norme ISO 13485. C’est pourquoi il est fortementrecommandé de choisir des fournisseurs eux-mêmes certifiés.
Les exigences relatives à la documentation du SMQ sont particulièrementnombreuses. Tout ce qui est fait dans l’entreprise doit être consciencieu-sement noté et conservé afin de servir de preuve de conformité au mo-ment de l’audit de certification. L’audit de certification est le contrôleréalisé par l’organisme notifié qui lui permet de vérifier que toutes lesexigences de la norme ISO 13485 (ou de l’annexe II de la directive93/42/CEE) sont mises en place et documentées dans l’entreprise.
La documentation du SMQ doit comprendre :
• Un manuel qualité
• Une politique qualité
• Des objectifs qualité
• Les procédures obligatoires mentionnées dans la norme
• Les spécifications des produits
• Les formulaires et autres documents nécessaires à l’entreprisepour fonctionner.
Cette documentation doit être vérifiée et approuvée, régulièrement miseà jour et sa diffusion doit êtremaîtrisée car elle sert de support de travailà chaque collaborateur. Une fois obsolètes, ces documents doivent êtreconservés pour démontrer que les dispositifs médicaux, plus anciens,produits à partir de ces documents sont conformes.
8.1.2 Responsabilité de la direction (§5)
En installant un système de management et en le faisant certifier, ladirection de l’entreprise prend des engagements vis-à-vis de ce système,de ses collaborateurs et de l’organisme notifié. Elle doit manifester cetengagement par un document écrit mais aussi dans ces actes. Sesengagements sont notamment :
• La planification du SMQ
• Le maintien de l’efficacité du SMQ
• La communication à ses collaborateurs de l’importance de respec-ter les dispositions du SMQ
• La rédaction et la mise à jour de la politique qualité
• La définition des objectifs qualité et leur planification
• La mise à disposition des ressources (humaines et matérielles)
• L’écoute du client
• La communication interne
• La délégation de l’autorité et de la responsabilité aux responsablesdes processus
• La réalisation de revues de direction régulières.
Les revues de direction sont le cœur du système de management de laqualité. Elles permettent de faire le point sur l’ensemble du SMQ et d’endéduire les actions à mettre en œuvre pour l’améliorer. Le rôle de la di-rection est primordial car c’est elle qui prend les décisions d’actions pourle SMQ, actions sans lesquelles le SMQ ne peut pas vivre (voir chapitre§8.1.5).
9Les numéros de paragraphe indiqués entre parenthèse font référenceaux numéros des paragraphes de la norme ISO 13485.

69Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE68
8.1.3 Management des ressources (§6)
L’entreprise doit assurer que les ressources disponibles sont suffisantespour mettre en œuvre le système de management de la qualité et enmaintenir l’efficacité. En particulier, elle doit s’assurer que chaque per-sonne est compétente à son poste de travail afin d’assurer la conformitédu produit. La compétence d’un collaborateur peut se démontrer par sonexpérience professionnelle, sa formation initiale ou des formationsinternes réalisées par l’entreprise. La preuve de chaque formation doitdonc être conservée et l’efficacité de la formation doit, en sus, êtredémontrée.
Du côté des ressources matérielles (infrastructures, équipements),celles-ci doivent être entretenues de façon à assurer la conformité duproduit. Les activités de maintenance doivent être définies. Dans le casparticulier des dispositifs médicaux nécessitant un environnement deproduction contrôlé (du type salle blanche ou flux laminaire, par exem-ple), ces installations doivent êtremaintenues et contrôlées comme touteressource matérielle.
8.1.4 Réalisation du produit (§7)
8.1.4.1 Planification de la réalisation du produit (§7.1)
La réalisation du produit, de l’achat des matières premières à l’expé-dition chez le client, doit être planifiée. Les flux de production doiventêtre décrits en cohérence avec les besoins du dispositif médical et desdifférents processus de l’entreprise. Il est nécessaire de décrire la sé-quence des étapes de production afin d’assurer qu’un produitconforme sera toujours disponible selon les exigences du client.
Par exemple, les flux de production peuvent être organisés demanièreà disposer en permanence d’unmois de stock de dispositifsmédicauxprêts à être envoyés ou de manière à produire un dispositif médicalconforme dans les délais définis avec le client. Quelle que soit lasolution retenue, elle doit être détaillée dans le SMQavec la planificationdes étapes qui en découlent.
La planification de la réalisation du produit doit inclure la gestion desrisques tout au long de la vie du produit. Cette gestion des risques peutêtre mise en place conformément à la norme harmonisée ISO 14971.
8.1.4.2 Processus relatifs aux clients (§7.2)
Le système de management de la qualité selon l’ISO 13485 a deuxobjectifs : d’une part la satisfaction des exigences réglementaires etd’autre part, la satisfaction des exigences du client. Tous les moyenspossibles doivent, de ce fait, être mis en œuvre pour que le client soitsatisfait. Il faut donc en premier lieu déterminer les exigences duclient et surtout les conserver comme preuve.
Le fabricant doit également vérifier qu’il peut répondre à l’ensembledes exigences du client avant d’accepter une commande : cette actions’appelait anciennement la « revue de contrat », et bien que cettedénomination ne soit plus mentionnée dans la norme, celle-ci estencore fréquemment utilisée.
Le système demanagement de la qualité doit contenir les dispositionsde communication avec le client :
• Comment le fabricant communique-t-il au client les informationsrelatives aux produits, aux services, au traitement de commandes…?
• Comment le fabriquant recueille-t-il et traite-t-il les retours desclients (réclamation, incidents…) ?
8.1.4.3 Conception et développement (§7.3)
Les exigences concernant la conception et le développement des dis-positifs médicaux sont très nombreuses, ceci afin d’assurer que leproduit développé sera conforme aux exigences essentielles de ladirective 93/42/CEE. La conception et le développement, incluantl’industrialisation, doit être planifiée. Cette planification inclut lesétapes de conception et de développement mais aussi les réunionsde revue de conception, les tests et essais à réaliser pour validerchaque étape et passer à la suivante ainsi que les responsabilités desdifférents acteurs du projet.
Un cahier des charges contenant les éléments suivants doit être crééet tenu à jour :

71Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE70
• Les exigences fonctionnelles, de performance et de sécurité
• Les exigences réglementaires et normatives applicables
• Les informations issues de conceptions similaires précédentes
• Une analyse des risques
• Toute autre exigence essentielle pour la conception et le développe-ment.
En sortie de conception, un dossier de conception doit être rédigéavec, entre autre, les spécifications d’achat des matières premières,les procédures de production et les critères d’acceptation auxcontrôles afin que la ligne de production du produit puisse être faci-lement intégrée dans le système de management de la qualité.
Toute modification d’un produit après son marquage CE doit se faireselon les même exigences que le développement d’un nouveau pro-duit afin d’assurer que le dispositif médical modifié sera toujoursconforme aux exigences essentielles de la directive.
8.1.4.4 Achats (§7.4)
Le processus Achats est un processus obligatoire de la norme. Sonobjectif est d’assurer que le « produit acheté » est conforme aux spé-cifications d’achats pour que le dispositif médical fabriqué avec le« produit acheté » soit également conforme. Il convient donc de définirdes spécifications d’achat pour chaque produit ou service à acheter.Elles comprennent la description du produit acheté mais aussi lescontrôles à effectuer sur le produit, les certifications requises pour lefournisseur et les procédures spécifiques à mettre en œuvre par lefournisseur.
A partir de ces spécifications d’achat, le fabricant sélectionne un four-nisseur capable de répondre à ces spécifications puis doit contrôler leproduit à réception. Il doit également évaluer régulièrement le four-nisseur pour vérifier qu’il continue à répondre aux spécificationsd’achat. La forme de cette évaluation (formulaire rempli par le fabri-cant, questionnaire au fournisseur, audit chez le fournisseur…)dépend de l’impact du produit acheté sur la qualité du produit fini etsur le client.
8.1.4.5 Production et préparation du service (§7.5)
Le système de management de la qualité doit comprendre les docu-ments de production décrivant :
• La planification de la production
• La validation des procédés de production
• La réalisation de la production incluant l’utilisation des équipements
• Le nettoyage et/ou la stérilisation
• Les contrôles en cours de production et le(s) contrôle(s) final(aux)
• Le conditionnement et l’étiquetage
• La libération du lot
• La livraison du produit
• Les prestations associées (maintenance, service après-ventes, ins-tallation, formation…)
• L’identification et la traçabilité
• La gestion de la propriété du client
• Le stockage
Cette documentation doit rester à disposition du personnel réalisantles opérations concernées. Elle doit être suffisamment détaillée pourqu’une personne non formée puisse effectuer la production d’un dis-positif médical conforme. Cette documentation étant confidentielle,il est important demettre en place lesmesures adéquates pour éviterqu’elle ne sorte de l’entreprise sans autorisation de la direction afind’éviter tout risque d’espionnage industriel.
8.1.4.6 Maîtrise des dispositifs de surveillance et de mesure (§7.6)
Les dispositifs de surveillance et demesure ou DSM sont des équipe-ments qui indiquent une mesure dont la valeur est nécessaire pourassurer la conformité du produit. Il s’agit, par exemple, des balances,thermomètres, règles graduées, affichages de température d’étuve,voltmètres, dosimètres. Ces DSMdoivent être régulièrement surveilléset étalonnés. Il est nécessaire de s’assurer qu’ils ne peuvent pas êtredéréglés et que leur incertitude de mesure ne remet pas en cause laconformité du dispositif médical.

73Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE72
Les étalons qui permettent l’étalonnage des DSM doivent être reliésaux étalons demesures internationaux. Si aucun étalon internationaln’existe pour la mesure, un étalon doit être conservé par l’entreprisedans un lieu qui en empêchera toute altération. Généralement, lesDSM sont vérifiés et étalonnés une fois par an tandis que les étalonssont vérifiés tous les 5 ans.
En sus des étalonnages annuels, il est conseillé de réaliser uncontrôle en vie fréquent sur les DSM (mise d’un poids étalon sur unebalance une fois par semaine, par exemple). En effet, lorsqu’uncontrôle ou un étalonnage fait apparaître une dérive sur un dispositifde mesure et de surveillance, la conformité de l’ensemble des pro-duits contrôlés avec ce DSM depuis le dernier contrôle est remise encause. Plus le contrôle est fréquent, plus le nombre de dispositifsmédicaux potentiellement non-conformes sera réduit et plus le coûtpour l’entreprise sera faible.
8.1.5 Mesures, analyse et amélioration (§8)
Le système de management de la qualité doit être constamment amé-lioré afin d’être bénéfique à l’entreprise. La surveillance comprend :
• Les retours d’information des clients (réclamations, incidents, non-conformités…)
• Les audits internes (audits réalisés par une entreprise sur sonpropre système afin d’en vérifier sa conformité à la norme et sonefficacité)
• Les indicateurs par processus (mesure chiffrée qui révèle l’efficacitéd’un processus : chiffre d’affaires mensuel, taux de rebuts de pro-duction, nombre de nouveaux clients par mois…)
• Les non-conformités des produits en cours de production ou aprèslivraison
L’objectif de cette surveillance est de détecter les anomalies, d’en définirles causes et de mettre en place les actions nécessaires pour que lesanomalies détectées ne se reproduisent plus. Il existe trois typesd’actions :
• Les actions correctives : elles corrigent la cause d’une anomalie dé-tectée afin que celle-ci ne se reproduise pas
• Les actions préventives : elles corrigent la cause d’une anomaliepotentielle afin que celle-ci ne se produise pas
• Les actions d’amélioration : elles permettent l’amélioration d’unprocessus sans qu’aucune anomalie n’ait été détectée (améliorationde la production par l’installation d’un nouvel équipement, parexemple).
Ces actions sont toujours mises en place dans un objectif précis et leurefficacité par rapport à l’objectif est évaluée. Si l’objectif n’est pas atteint,d’autres actions sontmises enœuvre jusqu’à ce que l’objectif soit atteint.
En plus du traitement des anomalies lors de leur survenue, des méta-analyses des données recueillies doivent être réalisées afin de recoupercertaines anomalies et d’engager des actions plus efficaces. Ce proces-sus de surveillance, d’analyse et d’actions permet l’amélioration encontinue du système demanagement de la qualité et donc sa rentabilité.
8.2 Annexe V : Assurance de la qualité de la production
Dans le cadre du marquage CE par l’annexe V de la directive 93/42/CEE, unsystème de management de la qualité doit être établi, mis en œuvre et cer-tifié par l’organisme notifié pour la production du dispositif médicalconcerné. Il s’agit d’un système de management de la qualité partiel, enparticulier, les paragraphes suivants de la norme ISO 13485 ne sont pasd’application :
• §5.2 Ecoute client
• §5.3.3 Communication interne
• §7.2 Processus relatifs aux clients
• §7.3 Conception et développement.
8.3 Annexe VI : Assurance de la qualité des produitsDans le cadre du marquage CE par l’annexe VI de la directive 93/42/CEE,un système de management de la qualité doit être établi, mis en œuvre etcertifié par l’organisme notifié pour la production du dispositif médicalconcerné. Il s’agit d’un système de management de la qualité partiel, enparticulier, les paragraphes suivants de la norme ISO 13485 ne sont pasd’application :

75Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE74
• §5.2 Ecoute client
• §5.3.3 Communication interne
• §6.3 Infrastructure
• §6.4 Environnement de travail
• §7.2 Processus relatifs aux clients
• §7.3 Conception et développement
• §7.4 Achats
• §7.5.2 Validation des processus de production et de la préparation duservice
• §7.5.4 Propriété du client.
L’objectif d’un SMQ selon l’annexe VI n’est pas d’assurer la maîtrise de laproduction mais simplement d’assurer la conformité du produit fini.
9 - LES ANNEXES III, IV ET VIII
9.1 Annexe III de la directive 93/42/CEE
L’annexe III ou « examen CE de type » consiste en la validation par l’orga-nisme notifié d’un (ou plusieurs) échantillon(s) représentatif(s) de la pro-duction appelé(s) « type ». L’évaluation se fait sur la base du dossiertechnique de marquage CE et d’essais réalisés soit par l’organisme notifié,soit par le fabricant chez un prestataire reconnu par l’organisme notifié.Dans tous les cas, le coût de l’évaluation et des essais supplémentaires està la charge du fabricant.
Si le fabricant revendique l’application de normes harmonisées pour établirla conformité de son dispositif médical aux exigences essentielles, l’orga-nisme notifié peut effectuer des audits sur site pour vérifier l’applicationde ces normes. Si l’évaluation de l’échantillon (du type) est satisfaisante,l’organisme notifié établit un certificat de conformité à l’annexe III pourcelui-ci. Cette annexe est toujours utilisée en combinaison avec une autreannexe afin de démontrer que l’ensemble de la production est identique autype validé et donc conforme.
9.2 Annexe IV de la directive 93/42/CEE
L’annexe IV ou « vérification CE » consiste en l’évaluation par l’organismenotifié de chaque lot de production. Les essais définis conjointement entrele fabricant et l’organisme notifié sont effectués (ou vérifiés) par l’organismenotifié. Ceux-ci peuvent être réalisés individuellement ou statistiquementen fonction du produit concerné. Généralement, pour les produits de grandesérie, seuls des contrôles statistiques sont effectués. L’organisme notifiédélivre un certificat pour chaque produit ou lot dont l’évaluation est satis-faisante.
A l’usage, cette procédure peut s’avérer onéreuse pour le fabricant car elleimplique l’intervention de l’organisme notifié pour chaque lot de fabricationet donc une facturation pour chaque intervention. L’annexe IV est utiliséepour les produits de très grande série dont la production s’effectue parcampagne (2 ou 3 lots par an) ou pour les produits très spécifiques du type

77Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE76
équipement dont moins de 10 pièces sortent de production chaque année.
En outre, le fabricant doit mettre en œuvre toutes les mesures nécessairespour que le procédé de fabrication assure la conformité du produit. Celan’implique pas la mise en place d’un système demanagement de la qualitémais des procédures de fabrication incluant les contrôles et leurs critèresd’acceptation doivent être écrites et respectées.
9.3 Les dispositifs médicaux sur-mesure
La directive 93/42/CEE définit les dispositifs médicaux sur-mesure comme :
« Tout dispositif fabriqué spécifiquement suivant la prescription écrite d'unpraticien dûment qualifié indiquant, sous la responsabilité de ce dernier,les caractéristiques de conception spécifiques et destiné à n'être utilisé quepour un patient déterminé.
La prescription susmentionnée peut également être établie par touteautre personne qui, en vertu de ses qualifications professionnelles, y estautorisée.
Les dispositifs fabriqués suivant des méthodes de fabrication continue ouen série qui nécessitent une adaptation pour répondre à des besoins spé-cifiques du médecin ou d'un autre utilisateur professionnel ne doivent pasêtre considérés comme des dispositifs sur mesure ».
Exemple de dispositifs médicaux sur-mesure :
• Prothèses dentaires
• Orthèses sur mesures (bas de contention, ceintures lombaires…)
• Semelles orthopédiques
Les dispositifs médicaux sur-mesure ne portent pas le marquage CE. Ilsdoivent cependant être conformes aux exigences essentielles de la direc-tive 93/42/CEE comme l’ensemble des dispositifs médicaux. Le fabricantdoit rédiger pour chaque dispositif mis sur le marché une déclarationcomprenant :
• Son nom et son adresse
• Les données permettant d'identifier le dispositif
• Une déclaration selon laquelle le dispositif est destiné à l'usageexclusif d'un patient déterminé et le nom de celui-ci
• Le nom de la personne qualifiée qui a établi la prescription et, le caséchéant, le nom de l'institution médicale concernée
• Les caractéristiques spécifiques du produit comme indiqué sur laprescription
• Une déclaration selon laquelle le dispositif est conforme aux exigencesessentielles de l'annexe I de la directive 93/42/CEE.
De plus, le fabricant s’engage à tenir à la disposition des autorités compé-tentes un dossier permettant de démontrer que le dispositif sur-mesureest effectivement conforme aux exigences essentielles. Ce dossier a unecomposition similaire à celle du dossier technique de marquage CE bienqu’il soit allégé car, si le dispositif sur-mesure est composé de produitsmarqués CE, leur conformité n’a pas à être démontrée à nouveau.
9.4 Les DM destinés à des investigations cliniques
La définition des « dispositifs médicaux destinés à des investigationscliniques » donnée par la directive 93/42/CEE est :
« Tout dispositif destiné à être mis à la disposition d'un médecin dûmentqualifié en vue de faire l'objet des investigations visées à l'annexe X point 2.1et effectuées dans un environnement clinique humain adéquat.
Aux fins de la réalisation des investigations cliniques, est assimilée aumédecin dûment qualifié toute autre personne qui, en vertu de ses qualifi-cations professionnelles, est autorisée à effectuer ces investigations ».
Ce paragraphe ne s’applique qu’aux dispositifs médicaux destinés auxétudes cliniques pré-marquage CE, c'est-à-dire, aux dispositifs médicauxn’ayant pas encore le marquage CE et dont l’étude clinique est nécessairepour l’obtenir.
Ces dispositifs doivent être conformes aux exigences essentielles de ladirective 93/42/CEE excepté celles pour lesquelles l’étude clinique estnécessaire. Le fabricant doit rédiger pour le(s) lot(s) de dispositifs mis àdisposition des praticiens une déclaration comprenant :
• Les données permettant d'identifier le(s) lot(s) de dispositifs
• Le protocole d’investigation clinique
• La brochure investigateur
• L'attestation d'assurance pour l’étude clinique

79Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE78
• Les documents utilisés pour obtenir le consentement éclairé despatients
• Une déclaration indiquant si le dispositif incorpore une substancedérivée du sang humain
• Une déclaration indiquant si le dispositif est fabriqué ou non à partir detissus d'origine animale
• L'avis délivré par le comité éthique concerné
• Le nom des praticiens et des institutions participant à l’étude clinique
• Les dates de début et de fin des investigations
•Une déclaration établissant que le(s) dispositif(s) est (sont) conforme(s)aux exigences essentielles, à l'exception des aspects faisant l'objet desinvestigations et que toutes les précautions ont été prises pour proté-ger la santé et la sécurité du patient.
Cette déclaration est conservée par le fabricant et mise à disposition desautorités compétentes. De plus, elle doit être jointe au dossier de demanded’autorisation d’investigation clinique si celle-ci porte sur des produits déjàfabriqués.
De plus, le fabricant doit réaliser un dossier technique de marquage CEpartiel comportant l’ensemble des éléments nécessaires au marquage CEà l’exception des points qui font l’objet des investigations cliniques. Ce dos-sier doit être tenu à la disposition des autorités compétentes pendant toutela durée des investigations cliniques puis pendant 5 ans après leur arrêt(15 ans dans le cas des dispositifs implantables).
9.5 Les DM de classe Is ou ImLes dispositifs médicaux de classe I stériles (Is) ou présentant une fonctionde mesurage (Im) doivent être évalués par un organisme notifié pour obte-nir le marquage CE. Cette évaluation ne porte que sur l’obtention et lemaintien de l’état stérile ou sur la fonction de mesurage (reproductibilité,maintenance, exactitude…).
Cette évaluation peut être documentaire ou faire l’objet d’un audit selonl’organisme notifié et l’organisation du fabricant. Il est essentiel pour le fa-bricant de produits de classe Is ou Im de prendre contact avec différentsorganismes notifiés afin de trouver le meilleur compromis entre les habi-tudes de l’organisme notifié et sa propre volonté.
10 - LES ASPECTS RÉGLEMENTAIRES À NE PASOUBLIER
10.1 La matériovigilance
Lorsqu’un fabricant appose le marquage CE sur son dispositif médical, ils’engage à respecter la législation concernant la matériovigilance. C'est-à-dire qu’il s’engage à recueillir, analyser, enregistrer et déclarer, si néces-saire, tout incident ou risque d’incident survenu à l’utilisation de sondispositif médical même si l’incident résulte d’un mésusage du produit.
Cette procédure de matériovigilance doit être intégrée dans le système demanagement de la qualité si un tel système est mis en place ou doit êtreintégrée dans le dossier technique demarquage CE. Le fabricant doit déclarerauprès de l’AFSSaPS un « correspondant de matériovigilance » et au moinsun suppléant. Ces deux personnes seront les correspondants privilégiés del’AFSSaPS en cas de déclaration d’incident réalisée par un établissement desanté ou un professionnel de santé.
Le suivi de la matériovigilance oblige le fabricant à mettre en place lesactions correctrices adéquates afin qu’un incident ne se reproduise pas ouqu’un risque d’incident ne se produise pas.
10.2 La Post-Market surveillance
La « Post Market Surveillance » ou « PMS » ou « surveillance aprèsmise surle marché » implique le recueil et l’analyse de toutes les données ayant unlien direct ou indirect avec le dispositif médical mis sur le marché afin d’in-tégrer ces données dans l’analyse des risques et de mettre en œuvre lesactions correctives, préventives ou d’amélioration en résultant.
Les données de PMS recouvrent :
• Les incidents ou risques d’incidents survenus à l’utilisation de produitssimilaires au dispositif concerné
• Les produits nouvellement mis sur le marché pour les mêmes indica-tions et présentant des performances supérieures au dispositifconcerné
• Les résultats d’études cliniques réalisées avec le dispositif

81Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE80
• Les données bibliographiques publiées sur la pathologie traitée et lesproduits ayant les mêmes indications.
Ces données sont recueillies et analysées par le fabricant d’un dispositifmédical et permettent de mettre à jour régulièrement le dossier technique,notamment les parties d’analyse des risques et d’évaluation clinique. Ellesdoivent permettre de détecter tout défaut de conception d’un produit pouvantnuire à ses performances ou à sa sécurité et ainsi permettre que seuls desdispositifs médicaux performants et sans risque soient présents sur lemarché.
10.3 La loi anti-cadeaux
Les produits de santé (médicaments et dispositifs médicaux) rembourséspar le système général de sécurité sociale en France sont soumis à une loispécifique dite « loi anti-cadeaux » ou loi « DMOS10». Cette loi interdit auxfabricants de produits de santé remboursés d’offrir des cadeaux en natureou en espèce aux prescripteurs de ces produits relevant d’un conseil de l’ordre(médecins, infirmiers, pharmaciens…). Cette disposition a été prise afind’éviter l’achat de prescriptions par un fabricant de produits remboursés.
Cette interdiction impose une procédure spécifique de demande d’avis auconseil de l’ordre du professionnel de santé concerné, lorsqu’un fabricantdésire inviter un praticien à un congrès ou à une manifestation marketing.Il est important de prendre en compte cette loi et de l’intégrer très tôt auxpratiques de l’entreprise car le responsable de l’entreprise est pénalementresponsable de tout manquement à cette loi.
Accessoire :Tout article qui, bien que n'étant pas un dispositif, est des-tiné spécifiquement par son fabricant à être utilisé avecun dispositif pour permettre l'utilisation dudit dispositifconformément aux intentions du fabricant de ce dispositif.
Afssaps :Agence Française de Sécurité Sanitaire des Produits deSanté. C’est l’autorité compétente Française pour les dis-positifs médicaux.
Analyse des éléments finis :Méthode d’analyse sur ordinateur qui permet de calculernumériquement le comportement d'objets (même trèscomplexes) soumis à diverses contraintes en représentantl’objet comme un ensemble fini d’éléments simples.
Analyse des risques :Correspond au recueil et à l’analyse critique de l’ensembledes phénomènes dangereux pouvant se produire à l’utili-sation du dispositif médical, des dangers associés à cesphénomènes dangereux et des actions à mettre en œuvrepour diminuer les risques. L’objectif de l’analyse desrisques est d’évaluer le rapport bénéfices / risques dudispositif.
10DMOS : Diverses Mesures d’Ordre Social
11 - GLOSSAIRE
Ce glossaire a pour objectif de donner une définition aux notions et termesspécifiques aumarquage CE des dispositifs médicaux utilisés dans ce guide.
A

83Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE82
Autocertification :Méthode demarquage CE des dispositifs médicaux de classe Idans laquelle le fabricant réalise son dossier technique demarquage CE puis déclare que son produit est conforme auxexigences essentielles de la directive 93/42/CEE sans l’inter-vention d’un organisme notifié.
Autorité compétente :Autorité nationale en charge du contrôle pour les dispositifsmédicaux. La mission de l’autorité compétente est d’assurerla sécurité d'emploi, l'efficacité, la qualité et le bon usage desdispositifs médicaux. En France, l’autorité compétente estl’Afssaps.
Carcinogénicité :Une substance carcinogène est une substance susceptible deprovoquer un cancer chez le patient.
Certification de produits :Procédure par laquelle un organisme certificateur atteste quele produit fabriqué par une entreprise est conforme à unenorme. La certification d’un produit garantit la qualité du pro-duit.
Certification d’entreprise :Procédure par laquelle un organisme certificateur atteste queles pratiques ou l’organisation d’une entreprise sont conformesà une norme appelée « référentiel de certification ». La vérifi-cation de l’organisme certificateur se réalise par un « audit »au sein de l’entreprise. La certification d’entreprise est unedémarche volontaire.
Commission Européenne :Autorité suprême pour la réglementation des dispositifs mé-dicaux dans l’Union Européenne.
Diagramme des flux :Représentation schématique des étapes nécessaires à lafabrication du dispositifmédical (achat et contrôle desmatièrespremières, usinage, assemblage, nettoyage, emballage,stérilisation…) auxquelles est associée une description desétapes de contrôle incluant leurs critères d’acceptation.
Directive :Texte de réglementation européen. La directive applicable auxdispositifs médicaux est la directive 93/42/CEE.
Dispositif médical :Tout instrument, appareil, équipement, logiciel, matière ouautre article, utilisé seul ou en association, y compris le logi-ciel destiné par le fabricant à être utilisé spécifiquement à desfins diagnostic et/ou thérapeutique, et nécessaire au bonfonctionnement de celui-ci, destiné par le fabricant à êtreutilisé chez l'homme à des fins :
- de diagnostic, de prévention, de contrôle, de traitementou d'atténuation d'une maladie,
- de diagnostic, de contrôle, de traitement, d'atténuationou de compensation d'une blessure ou d'un handicap,
- d'étude ou de remplacement ou modification de l'anato-mie ou d'un processus physiologique,
- de maîtrise de la conception,
et dont l'action principale voulue dans ou sur le corps humainn'est pas obtenue par des moyens pharmacologiques ou im-munologiques ni parmétabolisme, mais dont la fonction peutêtre assistée par de tels moyens.
C
D

85Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE84
Dispositif médical actif :Tout dispositif médical dépendant pour son fonctionnementd'une source d'énergie électrique ou de toute autre sourced'énergie que celle générée directement par le corps humainou la pesanteur.
Dispositif médical de diagnostic in vitro :Tout dispositif médical qui consiste en un réactif, un produitréactif, un matériau d'étalonnage, un matériau de contrôle,une trousse, un instrument, un appareil, un équipement ouun système, utilisé seul ou en combinaison, destiné par lefabricant à être utilisé in vitro dans l'examen d'échantillonsprovenant du corps humain, y compris les dons de sang et detissus, uniquement ou principalement dans le but de fournirune information :
- concernant un état physiologique ou pathologique
ou
- concernant une anomalie congénitale
ou
- permettant de déterminer la sécurité et la compatibilitéavec des receveurs potentiels
ou
- permettant de contrôler des mesures thérapeutiques.
Les récipients pour échantillons sont considérés comme desdispositifs médicaux de diagnostic in vitro. On entend par«récipients pour échantillons» des dispositifs, qu'ils soientsous vide ou non, spécifiquement destinés par leur fabricantà recevoir directement l'échantillon provenant du corps humainet à le conserver en vue d'un examen de diagnostic in vitro.
Les produits destinés à des usages généraux en laboratoirene sont pas des dispositifs médicaux de diagnostic in vitro àmoins que, eu égard à leurs caractéristiques, ils soient spé-cifiquement destinés par leur fabricant à des examens dediagnostic in vitro.
Dispositifmédical destiné à des investigations cliniques :Tout dispositif destiné à êtremis à la disposition d'unmédecindûment qualifié en vue de faire l'objet des investigationsvisées à l'annexe X point 2.1 et effectuées dans un environne-ment clinique humain adéquat.
Aux fins de la réalisation des investigations cliniques, estassimilée au médecin dûment qualifié toute autre personnequi, en vertu de ses qualifications professionnelles, est autoriséeà effectuer ces investigations.
Dispositif médical implantable actif :Tout dispositif médical actif qui est conçu pour être implantéen totalité ou en partie, par une intervention chirurgicale oumédicale, dans le corps humain ou, par une interventionmédicale, dans un orifice naturel et qui est destiné à resteraprès l'intervention.
Dispositif médical sur mesure :Tout dispositif fabriqué spécifiquement suivant la prescriptionécrite d'un praticien dûment qualifié indiquant, sous la res-ponsabilité de ce dernier, les caractéristiques de conceptionspécifiques et destiné à n'être utilisé que pour un patientdéterminé. La prescription susmentionnée peut égalementêtre établie par toute autre personne qui, en vertu de ses qua-lifications professionnelles, y est autorisée.
Les dispositifs fabriqués suivant desméthodes de fabricationcontinue ou en série qui nécessitent une adaptation pourrépondre à des besoins spécifiques dumédecin ou d'un autreutilisateur professionnel ne doivent pas être considéréscomme des dispositifs sur mesure.
Documents d’accompagnement :Documents accompagnant le dispositif médical, c'est-à-direla notice d’utilisation, le mode d’emploi, le conditionnement,l’étiquetage, la documentationmarketing, les fiches produits…

87Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE86
Dossier technique de marquage CE :Dossier contenant l’ensemble des spécifications et caracté-ristiques du produit et dont l’objectif est de démontrer laconformité du dispositif médical aux exigences essentiellesde l’annexe I de la directive 93/42/CEE. L’élaboration d’un dos-sier technique de marquage CE est obligatoire, quelles quesoient la classe du dispositif à marquer CE et la méthode demarquage CE choisie.
Essai de biocompatibilité :Essais biologiques réalisés sur des cellules (in vitro) ou surdes animaux (in vivo) permettant de démontrer que le produitinduit la réponse biologique voulue pour son utilisation (pro-lifération de cellules épithéliales, agrégation du sang…) etqu’il n’est pas toxique pour le patient.
Essai de fatigue :Essai mécanique consistant à soumettre le dispositif médicalaux contraintesmécaniques auxquelles il sera soumis durantson utilisation (pression, traction, torsion…) demanière répé-tée afin de s’assurer que celui-ci ne se détériorera pas àl’usage.
Evaluation clinique :Analyse des données issues de la bibliographie (revue desdonnées cliniques) ou d’une étude clinique (investigationclinique) permettant d’évaluer le comportement d’un dispositifmédical en utilisation clinique.
Exigences essentielles :Exigences de performance, de sécurité, de santé et deprotection de l’environnement énoncées dans les annexes desdirectives « nouvelle approche ». Ce sont les exigences
minimum auxquelles doivent répondre les produits pourpouvoir prétendre au marquage CE et donc être mis sur lemarché sur le territoire de l’Union Européenne.
Fabricant :La personne physique ou morale responsable de la concep-tion, de la fabrication, du conditionnement et de l'étiquetaged'un dispositif en vue de samise sur le marché en son proprenom, que ces opérations soient effectuées par cette mêmepersonne ou pour son compte par une tierce personne.
Et
La personne physique ou morale qui assemble, conditionne,traite, remet à neuf et/ou étiquette un ou plusieurs produitspréfabriqués et/ou leur assigne la destination d'un dispositifen vue de sa mise sur le marché en son nom propre.
Flow-chart de fabrication :Voir diagramme des flux de fabrication.
Investigation clinique :Etude clinique réalisée avec un dispositif médical (ou toutautre produit de santé) afin d’en évaluer l’innocuité et les per-formances chez l’homme en utilisation clinique. L’investiga-tion clinique se nomme également : recherche biomédicale.
ISO 13485 :Norme harmonisée de la directive 93/42/CEE concernantl’organisation du système de management de la qualité(SMQ). Cette norme décrit les exigences d’organisationnécessaires pour obtenir un SMQ conforme à la directive. Elleest écrite sur la base de l’ISO 9001 intégrant, en sus, lesexigences spécifiques aux dispositifs médicaux.
EF
I

89Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE88
Mandataire :Toute personne physique ou morale établie dans la Commu-nauté qui, après avoir été expressément désignée par le fabri-cant, agit et peut être contactée par les autorités et lesinstances dans la Communauté en lieu et place du fabricanten ce qui concerne les obligations que la directive« 93/42/CEE » impose à ce dernier.
Marquage CE :Symbole signifiant « Communauté Européenne ». Il est ap-posé sur les produits satisfaisants aux exigences essentiellesdes directives européennes dont ils relèvent. Il autorise lalibre circulation du produit dans les étatsmembres de l’UnionEuropéenne. Le marquage CE désigne également la procé-dure suivie par un fabricant pour obtenir le droit d’apposer lesymbole CE sur son produit.
Marquage CE médical :Le terme de marquage « CE médical » est un abus delangage. Il désigne lemarquage CE d’un produit conformémentà la directive 93/42/CEE relative aux dispositifs médicaux.
Matériovigilance :Procédure qui consiste à recueillir, analyser et déclarer(si besoin) l’ensemble des incidents ou risques d’incidentssurvenus lors de l’utilisation du dispositif médical.
Médicament :Toute substance ou composition présentée comme possédantdes propriétés curatives ou préventives à l'égard desmaladieshumaines. Toute substance ou composition pouvant être admi-nistrée à l'homme en vue d'établir un diagnosticmédical ou derestaurer, corriger ou modifier des fonctions physiologiqueschez l'homme est également considérée commemédicament.
Mutagénicité :Une substance mutagène est une substance susceptible deprovoque des mutations génétiques au cœur du noyau descellules du patient.
Norme :Recueil d’exigences, de méthodes et de spécificationsauxquelles un produit ou une organisation doit répondre pourattester de sa conformité à cette norme. Une norme estd’application volontaire.
Norme harmonisée :Norme qui définit les spécifications techniques, méthodesd’organisation et d’essais… nécessaires à un produit pour ré-pondre aux exigences essentielles d’une directive « nouvelleapproche ». Un produit conforme à une norme harmonisée aune présomption de conformité aux exigences essentielles dela directive correspondante.
Norme horizontale :Norme générale qui s’adresse à l’ensemble des produits d’undomaine.
Norme verticale :Norme spécifique qui s’adresse à un produit particulier ou àune catégorie de produits, définissant des exigences particu-lières pour ce produit ou ce type de produit et les méthodesd’essais permettant demesurer la conformité à ces exigencesparticulières.
Nouvelle approche :Démarche ayant pour objectif d’harmoniser la réglementation
MN

91Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE90
dans les étatsmembres de l’Union Européenne afin d’assurerla libre circulation des biens et desmarchandises. Le principede la nouvelle approche est de remplacer les réglementationsnationales par une réglementation européenne basée sur lesdirectives, ces directives énonçant un certain nombre d’exi-gences de performance et de sécurité pour les produits missur le marché.
Organisme notifié :Un organisme notifié (ou ON) est un organisme habilité parl’autorité compétente pour évaluer la conformité d’un dispo-sitif médical à la directive 93/42/CEE afin d’autoriser le fabri-cant à y apposer lemarquage CE. L’organisme notifié françaisest le LNE/Gmed.
Post-Market Surveillance ou PMS :Procédure qui consiste à recueillir, à analyser et à traiter lesinformations concernant les produits similaires présents surle marché et les données cliniques de la part du fabricant.Surveillance du marché pour l’autorité compétente.
Rapport bénéfices / risques :Ce rapport représente les avantages ou bénéfices apportéspar le dispositif médical par rapport aux risques encourus parle patient ou l’équipe médicale. Pour qu’un dispositif médicalpuisse être mis sur le marché, il est nécessaire que sonrapport bénéfices / risques soit positif, c'est-à-dire qu’ilapporte plus de bénéfices que de risques au patient.
Revue des données cliniques :Correspond au recueil et à l’analyse critique de l’ensembledes données cliniques (données bibliographiques ou issuesd’investigations cliniques) et dont l’objectif est d’évaluer lasécurité et les performances du dispositif médical et d’enidentifier les éventuels effets secondaires.
Système de management de la qualité ou SMQ :Forme précise d’organisation décrite dans des normes demanagement de la qualité et qui permet d’assurer que l’en-semble des produits fabriqués par l’entreprise répondent auxexigences des clients et aux exigences réglementaires.
Toxicité pour la reproduction :Une substance toxique pour la reproduction est une subs-tance susceptible de provoquer des malformations ou desmutations génétiques sur l’embryon, le fœtus ou l’enfant d’unpatient (homme ou femme) ayant été mis en en contact avecla substance.
O
P
R
S
T

93Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE92
12 - TABLE DES TABLEAUX
Tableau 1Liste des directives « nouvelle approche » 13
Tableau 2Exemple de tableau de classification (cas d’un appareilde radiodiagnostic conventionnel) 41
Tableau 3Choix de la méthode de marquage CE en fonction de la situation du fabricant 48
Tableau 4Exemples d’exigences essentielles et de normes harmonisées y répondant 53
Tableau 5Tableau d’évaluation de la conformité aux exigences essentielles 63
13 - TABLE DES FIGURES
Figure 1Schématisation de la notion de « norme verticale » et de « norme horizontale » 16
Figure 2Exemples de logo CE avec et sans intervention d’un organisme notifié 33
Figure 3Illustration de la notion de Fabricant au sens de la directive 34
Figure 4Procédures d’évaluation en fonction de la classe du dispositif médical 47
Figure 5La planification d’un projet de marquage CE 50
Figure 6Constitution d’un dossier de marquage CE 64

95Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE94
14 - ANNEXE 1 : LA DIRECTIVE 93/42/CEE
Cette annexe a pour objectif de présenter le plan de la directive 93/42/CEEafin d’aider le lecteur à trouver les informations qu’il recherche.
DIRECTIVE 93/42/CEE DU CONSEILdu 14 juin 1993 relative aux dispositifs médicaux
Article 1 - Définitions, champ d'applicationCet article regroupe l’ensemble des définitions nécessaires pour la compré-hension de la directive et limite son champ d’application aux dispositifs mé-dicaux et à leurs accessoires.
Article 2 - Mise sur le marché et mise en serviceCet article précise qu’un dispositif médical ne peut être mis à disposition del’utilisateur (à titre onéreux ou gratuit) que lorsqu’il satisfait à l’ensemble desexigences de la directive.
Article 3 - Exigences essentiellesCet article précise que les dispositifs médicaux doivent répondre auxexigences essentielles de l’annexe Imais aussi aux exigences essentielles desautres directives européennes qui leur sont applicables.
Article 4 - Libre circulation, dispositifs à destination particulièreCet article définit la libre circulation des dispositifs médicaux marqués CE,des dispositifs destinés aux investigations cliniques et des dispositifs sur-mesure. Il précise également que les états membres peuvent demander queles documents d’accompagnement soient rédigés dans leur langue nationale.
Article 5 - Renvoi aux normesCet article définit la notion de « norme harmonisée » et de présomption deconformité.
Article 6 - Comité «Normes et règles techniques»Cet article définit la création du comité supervisant la rédaction des normesharmonisées.
Article 7Cet article définit le rôle du comité « Normes et règles techniques ».

97Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE96
Article 8 - Clause de sauvegardeCet article définit la procédure à suivre pour les états membres qui veulentsuspendre la mise sur le marché, sur leur territoire national, d’un dispositifmédical marqué CE qui risque de compromettre la santé des utilisateurs. Ilprécise également les modalités de levée de la suspension ou, au contraire,d’extension à l’ensemble des étatsmembres, par la Commission Européenne,de la suspension de mise sur le marché, lorsque cela est nécessaire.
Article 9 - ClassificationCet article indique que la classification s’effectue en fonction des règles del’annexe IX. Il précise également les procédures à suivre en cas de désaccordentre le fabricant et son organisme notifié ou son autorité compétente.
Article 10 - Informations sur des incidents intervenus après la misedes dispositifs sur le marchéCet article définit les règles de matériovigilance applicables dans l’UnionEuropéenne pour les dispositifs médicaux.
Article 11 - Évaluation de la conformitéCet article définit les procédures de marquage CE des dispositifs médicauxen fonction de la classe de ceux-ci. Les procédures sont détaillées dans lesannexes II, III, IV, V, VI et VII auxquelles fait référence cet article.
Article 12 - Procédure spéciale pour les systèmes et nécessaires des-tinés à être assemblés en vue de constituer un dispositif et procédurede stérilisationCet article définit les procédures de marquage CE pour les fabricants quiassemblent des dispositifs médicaux marqués CE et pour les fabricants quistérilisent des dispositifs marqués CE, par dérogation à l’article 11.
Article 12 bis - Retraitement des dispositifs médicauxCet article prévoit la rédaction d’un rapport par la Commission Européennesur le retraitement des dispositifs médicaux.
Article 13 - Décisions enmatière de classificationet clause de dérogationCet article définit la procédure à suivre par les autorités compétenteslorsqu’elles veulent modifier une règle de classification ou ajouter une règlede dérogation à la classification pour certains produits.
Article 14 - Enregistrement des personnes responsablespour la mise sur le marché des dispositifsCet article définit la procédure de notification à l’autorité compétente des
responsablesde lamisesur lemarchédesdispositifsmédicauxetdumandatairedans l’Union Européenne.
Article 14 bis - Banque de données européenneCet article définit quelles sont les données présentes dans la base de donnéeseuropéenne des dispositifs médicaux à laquelle toutes les autorités compé-tentes ont accès ainsi que la procédure pour transmettre ces données.
Article 14 ter - Mesures particulières de veille sanitaireCet article définit la procédure permettant à un état membre d’interdire lamise sur lemarché d’un dispositif sur son territoire afin d’assurer la sécuritédes utilisateurs.
Article 15 - Investigations cliniquesCet article définit la procédure applicable pour la réalisation d’investigationscliniques sur les dispositifsmédicaux (en se référant à l’annexe X) ainsi que laprocédure pour la mise à disposition des dispositifs destinés à ces investiga-tions cliniques (non marqués CE).
Article 16 - Organismes notifiésCet article définit la procédure à suivre pour une autorité compétente afin denotifier et de déclarer un organisme notifié. Il précise également les obliga-tions des organismes notifiés vis-à-vis de leur autorité compétente et de laCommission Européenne.
Article 17 - Marquage CECet article précise comment doit être apposé le marquage CE sur les dispo-sitifs médicaux tout en se référant à l’annexe XII.
Article 18 - Marquage CE indûment apposéCet article définit les droits des états membres en cas de détection de mar-quage CE indûment apposé (marquage CE apposé sur un produit nonconforme ou fabricant n’ayant pas suivi la procédure de marquage CE).
Article 19 - Décisions de refus ou de restrictionCet article définit la procédure à suivre par l’autorité compétente, vis-à-vis dufabricant pour le refus, la restriction ou le retrait d’un dispositif médical surson territoire.
Article 20 - ConfidentialitéCet article définit les informations qui ne sont pas confidentielles et quipeuvent, à ce titre, être mises à la disposition de l’ensemble des autoritéscompétentes, voire du public.

99Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE98
Article 20 bis - CoopérationCet article introduit l’obligation, pour les étatsmembres, de coopérer entre eux.
Article 21 - Abrogation et modification de directivesCet article recense l’ensemble des directives modifiées et abrogées pourpermettre l’application de la directive 93/42/CEE.
Article 22 - Mise enœuvre, dispositions transitoiresCet article définit la date demise en application définitive de la directive et lesprocédures existantes pendant la période transitoire.
Article 23Cet article informe que tous les états membres sont destinataires de ladirective.
ANNEXE I - EXIGENCES ESSENTIELLESCette annexe liste l’ensemble des exigences essentielles auxquelles le dispo-sitif médical doit être conforme pour prétendre au marquage CE.Plan de l’annexe :I. Exigences généralesII. Exigences relatives à la conception et la construction• Propriétés chimiques, physiques et biologiques• Infection et contamination microbienne• Propriétés relatives à la fabrication et à l'environnement• Dispositifs ayant une fonction de mesurage• Protection contre les rayonnements• Exigences pour les dispositifsmédicaux raccordés à une source d'énergieou équipés d'une telle source
• Informations fournies par le fabricant
ANNEXE II - DÉCLARATION CE DE CONFORMITÉ(Système complet d'assurance de la qualité)Cette annexe définit les exigences relatives à la mise en place d’un Système
de management de la qualité complet ainsi que les exigences du dossier deconception.Plan de l’annexe :
• Obligations du fabricant• Système de qualité• Examen de la conception du produit• Surveillance• Dispositions administratives• Application aux dispositifs de la classe IIa et IIb
ANNEXE III - EXAMEN CE DE TYPELa procédure décrite dans l’annexe III est la procédure par laquelle l’orga-nisme notifié constate et atteste qu’un échantillon représentatif du dispositifmédical (le type) est conforme aux exigences de la directive 93/42/CEE.Plan de l’annexe :
• Définition de l’examen CE de type• Obligations du fabricant• Contenu de la documentation technique (dossier technique)• Mission de l’organisme notifié• Dispositions administratives
ANNEXE IV - VÉRIFICATION CELa procédure décrite dans l’annexe IV est la procédure par laquelle l’orga-nisme notifié constate et atteste que chaque production de dispositifs médi-caux est conforme aux exigences de la directive 93/42/CEE.Plan de l’annexe :
• Définition de la vérification CE• Obligations du fabricant• Vérification par contrôle et essai de chaque produit• Vérification statistique• Dispositions administratives• Application aux dispositifs de la classe IIa

101Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE100
ANNEXE V - DÉCLARATION CE DE CONFORMITÉ(Assurance de la qualité de la production)Cette annexe définit les exigences relatives à la mise en place d’un Systèmedemanagement de la qualité de la production du dispositif médical concerné.Plan de l’annexe :
• Obligations du fabricant• Système de qualité• Surveillance• Dispositions administratives• Application aux dispositifs de la classe IIa
ANNEXE VI - DÉCLARATION CE DE CONFORMITÉ(Assurance de la qualité des produits)Cette annexe définit les exigences relatives à la mise en place d’un Systèmede management de la qualité du dispositif médical.Plan de l’annexe :
• Obligations du fabricant• Système de qualité• Surveillance• Dispositions administratives• Application aux dispositifs de la classe IIa
ANNEXE VII - DÉCLARATION CE DE CONFORMITÉLa procédure décrite dans l’annexe VII est la procédure par laquelle le fabricantassure et déclare que les dispositifs médicaux qu’il fabrique sont conformesaux exigences de la directive 93/42/CEE. C’est l’ « autocertification ».Plan de l’annexe :
• Définition de la déclaration CE de conformité• Obligations du fabricant• Contenu de la documentation technique (dossier technique)• Application aux dispositifsmis sur lemarché à l’état stérile ou ayant unefonction de mesurage
• Application aux dispositifs de la classe IIa
ANNEXE VIII - DÉCLARATION RELATIVE AUX DISPOSITIFS AYANTUNE DESTINATION PARTICULIÈRECette annexe décrit les procédures à suivre pour mettre à disposition lesdispositifs médicaux destinés à des investigations cliniques et les dispositifsmédicaux fabriqués sur-mesure.Plan de l’annexe :
• Contenu de la déclaration pour les dispositifs sur-mesure• Contenu de la déclaration pour les dispositifs destinés aux investigationscliniques
• Documentation technique pour les dispositifs sur-mesure•Documentation technique pour les dispositifs destinés aux investigationscliniques
• Vigilance
ANNEXE IX - CRITÈRES UTILISÉS POUR LA CLASSIFICATIONCette annexe définit les règles permettant de classer les dispositifsmédicauxdans les quatre classes de risques (I, IIa, IIb, III).Plan de l’annexe :I. Définitions
• Durée• Dispositifs invasifs• Instrument chirurgical réutilisable• Dispositif actif thérapeutique• Dispositif actif destiné au diagnostic• Système circulatoire central• Système nerveux central
II. Règles d'application
III. Classification• Dispositifs non invasifs• Dispositifs invasifs• Autres règles applicables aux dispositifs actifs• Règles spéciales

103Guide du marquage CE des dispositifs médicaux selon la directive 93/42/CEE102
ANNEXE X - ÉVALUATION CLINIQUECette annexe définit la procédure à suivre pour réaliser l’évaluation clinique quicomprend l’analyse des données cliniques (données bibliographiques) et lesinvestigations cliniques (études cliniques, recherches biomédicales).Plan de l’annexe :
• Dispositions générales• Investigations cliniques
ANNEXE XI - CRITÈRES MINIMAUX DEVANT ÊTRE RÉUNIS POURLA DÉSIGNATION DES ORGANISMES À NOTIFIERCette annexe définit les critères de notification et d’évaluation des organismesnotifiés. Cette annexe n’est appliquée que par les autorités compétentes.
ANNEXE XII - MARQUAGE CE DE CONFORMITÉCette annexe donne le format du logo CE à appliquer sur les dispositifsmédi-caux conformes à la directive 93/42/CEE et aux autre directives applicables(le cas échéant).
15 - ANNEXE 2 : POUR PLUS D’INFORMATION…
Liste des normes harmonisées :http://www.newapproach.org/Directives/DirectiveList.asp
Liste des organismes notifiés :http://ec.europa.eu/enterprise/newapproach/nando/index.cfm?fuseac-tion=directive.main
Liste des autorités compétentes :http://ec.europa.eu/enterprise/medical_devices/ca/list_ca.htm
Site Internet de la section dispositifs médicaux de la CommissionEuropéenne :http://ec.europa.eu/enterprise/medical_devices/index_en.htm
Site Internet EurLex (accès à la réglementation européenne) :http://eur-lex.europa.eu/fr/index.htm
Site Internet Legifrance (accès à la réglementation française) :http://www.legifrance.gouv.fr/
Site Internet de l’Afssaps :http://agmed.sante.gouv.fr/
Site Internet de l’Afnor :http://www.afnor.fr/portail.asp
Site Internet du LNE / Gmed :http://www.gmed.fr/
Guides en ligne du MEDDEV :http://ec.europa.eu/enterprise/medical_devices/meddev/index.htm
Site Internet du CRITT Santé Bretagne :http://www.critt-sante.fr/
Site Internet de Twoksa Conseil :http://www.twoksa.com


















