DDoossssiieerr dduu CCNNHHIIMM - Bienvenue sur le site du ... du CNHIM - PDF/PDF 2010... · Sur...
68
Le CNHIM est une association indépendante à but non lucratif (loi 1901) dont la vocation est de réaliser et diffuser une information rigoureuse et scientifique sur le médicament. Tous les articles publiés dans Dossier du CNHIM sont le fruit d'un travail collectif, sur le fond et sur la forme, entre les rédacteurs signataires, le comité de rédaction, et la rédaction du CNHIM d'une part, le comité de lecture et certains experts, spécialistes du sujet traité, d'autre part. Sur chaque sujet, Dossier du CNHIM ne publie donc pas les opinions de tel ou tel, mais réalise une analyse scientifique critique, la plus objective possible. Malgré tout le soin apporté à l’élaboration de Dossier du CNHIM, une erreur peut se glisser dans les informations diffusées. Les lecteurs doivent donc conserver la plus grande vigilance dans l’exploitation des données à leur disposition. Directeur de la Publication : Xavier Dode Rédaction Rédactrice en chef : Marie Caroline Husson Comité de rédaction : Hélène Bourgoin-Hérard (Tours), Dominique Dardelle (Suresnes), Albert Darque (Marseille), Bérangère Gruwez (Berck), Isabelle Fusier (Paris), Isabelle Jolivet (Paris), Véronique Lecante (Paris), Nathalie Le Guyader (Paris), Corinne Tollier (Paris). Comité de lecture : A. Baumelou (Paris), P. Beaufils (Paris), P. Faure (Paris), J.E. Fontan (Paris) C. Guérin (Paris), S. Limat (Besançon), C. Montagnier-Pétrissans (Paris), M. Ollagnier (St Etienne), G. Vedel (Paris), J.M. Vetel (Le Mans), T. Vial (Lyon). Experts pour ce numéro : Isabelle Fusier, Aurore Gouraud, Marie-Caroline Husson, Hubert Marotte, Alain Saraux, Corinne Tollier. Rythme de parution : 6 numéros par an N° ISSN 0223.5242. N° de commission paritaire : G 82049 IMPRESSION : CPI – Imprimerie France Quercy ZA des Grands Camps, 46090 Mercuès. CENTRE NATIONAL HOSPITALIER D’INFORMATION SUR LE MEDICAMENT www.cnhim.org Hôpital de Bicêtre – 78, rue du Général Leclerc B.P. 11 - 94272 Le Kremlin Bicêtre cedex Tél. : 01 46 58 07 16 – Fax : 01 46 72 94 56 Courriel : [email protected] Président : Xavier Dode Président fondateur : André Mangeot Secrétariat abonnement : Zohra El Hadaoui Conseil d’Administration : F. Ballereau (Nantes), G. Benoit (Paris), H. Bontemps (Villefranche/Saône), O. Bourdon (Paris), E. Boury (Lomme), J. Calop (Grenoble), B. Certain (Paris), M. Courbard-Nicolle (Paris), D. Dardelle (Suresnes), X. Dode (Lyon), E. Dufay (Lunéville), J.E. Fontan (Bondy), D. Goeury (Paris), C. Guérin (Paris), M.C. Husson (Paris), J.F. Latour (Lyon), N. Le Guyader (Paris), A. Lepelletier (Nantes), P. Passe (Paris), P. Paubel (Paris), F. Pinguet (Montpellier), C. Rieu (Longjumeau), D. Roncalez (Colmar), J.M. Trivier (Lille). Echos du CNHIM Marie-Claude Saux, Frank Jorgensen 3 Polyarthrite rhumatoïde (2 ère partie) : nouvelles biothérapies ciblant les cytokines : tocilizumab, certolizumab, golimumab E. Radideau, S. Bah, C. Dupont, P. Hilliquin et la participation du comité de rédaction Editorial Hubert Marotte 5 En bref Marie-Caroline Husson 6 1. Introduction 7 2. Tocilizumab 8 2.1. Renseignements généraux et galéniques 8 2.2. Propriétés pharmacologiques 8 2.3. Efficacité clinique 9 2.4. Renseignements thérapeutiques 26 2.5. Plan de gestion des risques et cohortes de suivi 29 3. Certolizumab 30 3.1. Renseignements généraux et galéniques 30 3.2. Propriétés pharmacologiques 30 3.3. Efficacité clinique 31 3.4. Renseignements thérapeutiques 37 3.5. Plan de gestion des risques 40 4. Golimumab 40 4.1. Renseignements généraux et galéniques 40 4.2. Propriétés pharmacologiques 40 4.3. Efficacité clinique 40 4.4. Renseignements thérapeutiques 40 5. Stratégie thérapeutique dans la polyarthrite rhumatoïde 52 5.1. Biothérapies dans la polyarthrite rhumatoïde 52 5.2. Proposition de stratégie d’utilisation des biothérapies 52 Conclusion 54 Abréviations et acronymes 56 Références bibliographiques 56 Fiche Interactions Médicamenteuses Inhibiteurs de la phosphodiestérase 5 59 A. Hugon, I. Federspiel, P. Bedouch, B. Allenet, J. Calop Résumés des derniers numéros parus 65 Au sommaire de Dossier du CNHIM 66 Bulletin d’abonnement 2010 67 Dossier du CNHIM participe à l’ISDB, réseau international de revues indépendantes de formation thérapeutique. Le CNHIM a la propriété des textes publiés dans ce numéro et se réserve tous les droits de reproduction (même partielle), d’adaptation, de traduction, pour tous les pays et par quelque procédé que ce soit (loi du 11 mars 1957, art. 40 et 41 du Code Pénal art. 425). Les articles de Dossier du CNHIM sont indexés dans bibliopch. D D D o o o s s s s s s i i i e e e r r r d d d u u u C C C N N N H H H I I I M M M 2010 Tome XXXI, 5 E v a l u a t i o n F i c h e s
-
Upload
nguyendieu -
Category
Documents
-
view
213 -
download
0
Transcript of DDoossssiieerr dduu CCNNHHIIMM - Bienvenue sur le site du ... du CNHIM - PDF/PDF 2010... · Sur...

Le CNHIM est une association indépendante à but non lucratif (loi 1901) dont la vocation est de réaliser et diffuser une information rigoureuse et scientifique sur le médicament. Tous les articles publiés dans Dossier du CNHIM sont le fruit d'un travail collectif, sur le fond et sur la forme, entre les rédacteurs signataires, le comité de rédaction, et la rédaction du CNHIM d'une part, le comité de lecture et certains experts, spécialistes du sujet traité, d'autre part. Sur chaque sujet, Dossier du CNHIM ne publie donc pas les opinions de tel ou tel, mais réalise une analyse scientifique critique, la plus objective possible. Malgré tout le soin apporté à l’élaboration de Dossier du CNHIM, une erreur peut se glisser dans les informations diffusées. Les lecteurs doivent donc conserver la plus grande vigilance dans l’exploitation des données à leur disposition.
Directeur de la Publication : Xavier Dode
Rédaction
Rédactrice en chef : Marie Caroline Husson
Comité de rédaction : Hélène Bourgoin-Hérard (Tours), Dominique Dardelle (Suresnes), Albert Darque (Marseille), Bérangère Gruwez (Berck), Isabelle Fusier (Paris), Isabelle Jolivet (Paris), Véronique Lecante (Paris), Nathalie Le Guyader (Paris), Corinne Tollier (Paris). Comité de lecture : A. Baumelou (Paris), P. Beaufils (Paris), P. Faure (Paris), J.E. Fontan (Paris) C. Guérin (Paris), S. Limat (Besançon), C. Montagnier-Pétrissans (Paris), M. Ollagnier (St Etienne), G. Vedel (Paris), J.M. Vetel (Le Mans), T. Vial (Lyon). Experts pour ce numéro : Isabelle Fusier, Aurore Gouraud, Marie-Caroline Husson, Hubert Marotte, Alain Saraux, Corinne Tollier.
Rythme de parution : 6 numéros par an N° ISSN 0223.5242. N° de commission paritaire : G 82049 IMPRESSION : CPI – Imprimerie France Quercy ZA des Grands Camps, 46090 Mercuès.
CENTRE NATIONAL HOSPITALIER D’INFORMATION SUR LE MEDICAMENT
www.cnhim.org
Hôpital de Bicêtre – 78, rue du Général Leclerc B.P. 11 - 94272 Le Kremlin Bicêtre cedex
Tél. : 01 46 58 07 16 – Fax : 01 46 72 94 56 Courriel : [email protected]
Président : Xavier Dode Président fondateur : André Mangeot �
Secrétariat abonnement : Zohra El Hadaoui
Conseil d’Administration : F. Ballereau (Nantes), G. Benoit (Paris), H. Bontemps (Villefranche/Saône), O. Bourdon (Paris), E. Boury (Lomme), J. Calop (Grenoble), B. Certain (Paris), M. Courbard-Nicolle (Paris), D. Dardelle (Suresnes), X. Dode (Lyon), E. Dufay (Lunéville), J.E. Fontan (Bondy), D. Goeury (Paris), C. Guérin (Paris), M.C. Husson (Paris), J.F. Latour (Lyon), N. Le Guyader (Paris), A. Lepelletier (Nantes), P. Passe (Paris), P. Paubel (Paris), F. Pinguet (Montpellier), C. Rieu (Longjumeau), D. Roncalez (Colmar), J.M. Trivier (Lille).
Echos du CNHIM Marie-Claude Saux, Frank Jorgensen 3
Polyarthrite rhumatoïde (2ère partie) : nouvelles biothérapies ciblant les cytokines : tocilizumab, certolizumab, golimumab
E. Radideau, S. Bah, C. Dupont, P. Hilliquin et la participation du comité de rédaction
Editorial Hubert Marotte 5
En bref Marie-Caroline Husson 6
1. Introduction 7
2. Tocilizumab 8 2.1. Renseignements généraux et galéniques 8 2.2. Propriétés pharmacologiques 8 2.3. Efficacité clinique 9 2.4. Renseignements thérapeutiques 26 2.5. Plan de gestion des risques et cohortes de suivi 29
3. Certolizumab 30 3.1. Renseignements généraux et galéniques 30 3.2. Propriétés pharmacologiques 30 3.3. Efficacité clinique 31 3.4. Renseignements thérapeutiques 37 3.5. Plan de gestion des risques 40
4. Golimumab 40 4.1. Renseignements généraux et galéniques 40 4.2. Propriétés pharmacologiques 40 4.3. Efficacité clinique 40 4.4. Renseignements thérapeutiques 40
5. Stratégie thérapeutique dans la polyarthrite rhumatoïde 52 5.1. Biothérapies dans la polyarthrite rhumatoïde 52 5.2. Proposition de stratégie d’utilisation des biothérapies 52
Conclusion 54
Abréviations et acronymes 56
Références bibliographiques 56
Fiche Interactions Médicamenteuses
Inhibiteurs de la phosphodiestérase 5 59 A. Hugon, I. Federspiel, P. Bedouch, B. Allenet, J. Calop Résumés des derniers numéros parus 65 Au sommaire de Dossier du CNHIM 66 Bulletin d’abonnement 2010 67
Dossier du CNHIM participe à l’ISDB, réseau international de revues indépendantes de formation thérapeutique.
Le CNHIM a la propriété des textes publiés dans ce numéro et se réserve tous les droits de reproduction (même partielle), d’adaptation, de traduction, pour tous les pays et par quelque procédé que ce soit (loi du 11 mars 1957, art. 40 et 41 du Code Pénal art. 425). Les articles de Dossier du CNHIM sont indexés dans bibliopch.
DDDooossssssiiieeerrr ddduuu CCCNNNHHHIIIMMM 2010 Tome XXXI, 5
E
v
a
l
u
a
t
i
o
n
Fiches

-2-
Dossier du CNHIM, 2010, XXXI, 5

-3-
Dossier du CNHIM, 2010, XXXI, 5
Echos du CNHIM
La Société Française de Pharmacie Clinique (SFPC) et l’European Society of Clinical Pharmacy (ESCP) ont uni leurs forces pour partager, informer et former les pharmaciens cliniciens Français et Européens aux principales activités de la pharmacie clinique en organisant une manifestation commune. Le 13ème congrès de la Société Française de Pharmacie Clinique et le 39ème Symposium Européen de Pharmacie Clinique se déroulent donc conjointement à Lyon les 21, 22, 23 Octobre 2010. L’occasion m’est ainsi fournie de donner la parole à Marie-Claude Saux, Présidente de la SFPC, et à Frank Jorgensen, Président de l’ESCP. Je les en remercie très chaleureusement.
Marie-Caroline Husson Rédactrice en chef
Sachons défendre une information indépendante La Société Française de Pharmacie Clinique (SFPC) est engagée dans le « bon usage » par la promotion de l’utilisation sûre, efficace et rationnelle des produits de santé, et contribue à exercer une mission d’information scientifique sur ces produits pour les malades et les autres professionnels de santé.
L’intérêt prédominant de nos actions est celui du malade qui doit bénéficier de la meilleure prise en charge au meilleur coût pour la collectivité ; or il n’y a pas de « juste prescription » ni de maîtrise de la consommation sans maîtrise de l’information afférente.
Pour exercer son métier au sein des établissements de santé ou dans les officines de ville, le pharmacien clinicien doit pouvoir s’appuyer sur des informations claires, précises, indépendantes de tous lobbies et qui intègrent, au delà des données du RCP, l’ensemble des acquis scientifiques tels que données d’auteurs et avis d’experts.
Les outils produits par le CNHIM, la revue d’évaluation thérapeutique Dossier du CNHIM et la base Thériaque®, correspondent aux attentes du pharmacien et du médecin sur les médicaments depuis très longtemps, et plus récemment sur les dispositifs médicaux.
Thériaque® est le réceptacle de toutes les expertises en particulier hospitalières ; son niveau inégalé de structuration et de codification de l’information ouvre des perspectives pour le développement de fonctionnalités contribuant à la sécurisation du
E c h o s d u C N H I M
Marie-Claude Saux Hôpital du Haut-Lévêque, CHU Bordeaux, Service Pharmacie
Présidente de la Société Française de Pharmacie Clinique (SFPC)

-4-
Dossier du CNHIM, 2010, XXXI, 5
Echos du CNHIM
circuit du médicament dans les Logiciels d’Aide à la Prescription (LAP).
Thériaque® est entièrement pilotée par des confrères praticiens hospitaliers qui aspirent à construire une base répondant aux besoins des pharmaciens praticiens de terrain ; en facilitant leurs actions et en améliorant la qualité et leur efficience, Thériaque® se différencie des autres bases de données.
Au moment où les systèmes d’information hospitaliers arrivent à maturité, où les réseaux ville-hôpital demeurent une évidence au travers de nouveaux outils comme le Dossier Pharmaceutique, Thériaque® est la base idéale à laquelle tout pharmacien professionnel de santé doit avoir accès.
Soutenir le CNHIM en s’abonnant à la revue Dossier du CNHIM, et en privilégiant le choix de la base
Thériaque® dans son établissement ou en investissant du temps et de l’énergie pour sa constitution, est un acte citoyen au regard des enjeux actuels de la maîtrise des sources d’information dans les outils informatiques.
Le soutien à Thériaque® doit être fort devant l’arrivée de logiciels de plus en plus performants mettant à disposition des utilisateurs dans les outils d’aide à la décision une information « prête à consommer » souvent très influencée...
Maîtriser l’information est indispensable pour un exercice professionnel de qualité et indépendant. Thériaque® est une richesse collective et une opportunité pour ceux qui veulent disposer d’une information sure, fiable, riche, et libre.
Sachons la défendre, et en profiter.
La pharmacie clinique en Europe au front de l’innovation La pharmacie clinique est en plein développement à travers l'Europe et au-delà. Dans le même temps, le monde du médicament, des dispositifs médicaux et de la pharmacothérapie devient de plus en plus performant, coûteux et complexe. Une gestion réussie des thérapeutiques et de l'économie simultanément, dépend de la présence de professionnels hautement qualifiés et compétents, parmi eux les pharmaciens cliniciens.
Le choix du thème principal du 39ème Symposium d'automne de l'ESCP et 13ème Congrès de la SFPC (Lyon, 21-23 Octobre 2010), ”La pharmacie clinique au front de l’innovation”, reflète ce scénario, avec l'objectif pour l'ESCP d'être un foyer important de formation et d'apprentissage.
Dans les domaines des accidents vasculaires cérébraux, de certains types de cancers, et de l’insuffisance cardiaque, l’objectif a été de fournir aux participants les informations les plus récentes sur la façon de gérer ces maladies graves et répandues.
Grâce à de nombreux ateliers et à un nombre record de résumés acceptés (près de 500) pour publications affichées ou orales, l'occasion a été ainsi donnée d'informer sur les derniers développements de la pharmacie clinique dans la plupart des pays Européens et au-delà, au sein d'un large éventail de domaines de pratiques.
Des Dossiers d'actualité, organisée par le conseil des Special Interest Groups (SIG) de l'ESCP, ont permis de présenter les plus récentes innovations dans un certain nombre de domaines (pédiatrie, gériatrie, nutrition clinique, pharmacoéconomie etc...).
Enfin un accent particulier est mis actuellement sur les dispositifs médicaux. C'est l'un des résultats positifs de la collaboration entre l'ESCP et la SFPC, puisqu’en France les pharmaciens cliniciens sont fortement impliqués dans l'approvisionnement, la gestion et l'utilisation rationnelle des dispositifs médicaux.
Nous remercions les laboratoires BAYERHEALTHCARE
qui participent à l’impression de BMS
Dossier du CNHIM en 2010. SERVIER
Frank Jorgensen MSc in Pharmacy, ClinDipPharm, Hospital Pharmacy, PO 1, 5021 Bergen, Norway
Président de la Société Européenne de Pharmacie Clinique (ESCP)

-5-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
La polyarthrite rhumatoïde : l’ère des biothérapies
Cette année à la suite d'un travail collaboratif international, de nouveaux critères spécifiques de la polyarthrite rhumatoïde précoce ont été publiés conjointement dans les revues européennes et américaines de rhumatologie. Ces critères permettent maintenant de diagnostiquer précocement la maladie. Le but est de pouvoir débuter un traitement le plus rapidement possible avant l'apparition de la destruction articulaire et ainsi de prévenir son apparition. Ces critères étaient indispensables depuis l'apparition des biothérapies. Ces biothérapies, développées à partir d'une meilleure compréhension de la physiopathologie de la maladie, permettent d'améliorer la symptomatologie des malades et surtout de stopper la destruction articulaire. Cependant, leur utilisation n'est pas sans effet indésirable. Pour mémoire, il y a déjà 10 ans, la pharmacovigilance a identifié de nombreux cas de réactivation de tuberculose latente chez les malades traités par inhibiteur du TNFα (tumor necrosis factor alpha). Depuis trois ans, trois nouvelles biothérapies sont venues renforcer notre arsenal thérapeutique. La première est l'abatacept qui inhibe la co-stimulation. La deuxième est le tocilizumab qui est un anticorps se fixant sur le récepteur de l'interleukine-6. La dernière est le certolizumab pegol qui est un nouvel inhibiteur du TNFα sous forme pegylé. Devant la multiplication des biothérapies, il nous faut développer une prise en charge personnalisée
pour chaque malade. Pour cela, il est nécessaire de valider des biomarqueurs à la fois de sévérité de la maladie polyarthrite rhumatoïde, et de la réponse aux différentes biothérapies. Cela permettrait pour le malade de bénéficier de la biothérapie dont il a besoin, sans lui faire prendre le risque d'effet indésirable potentiellement mortel d'un médicament inefficace. Enfin, cela permettrait de réduire les coûts pour la collectivité avec un malade qui va mieux avec son traitement personnalisé sans la gestion des effets indésirables. Comme pour les pathologies chroniques, la prise en charge d’une polyarthrite rhumatoïde doit être multidisciplinaire. Ceci implique une information régulière et actualisée des professionnels de santé. Le traitement de fond de première intention reste le méthotrexate qui a un excellent profil de tolérance. Cette pathologie et ses traitements avaient déjà fait l’objet d’un Dossier du CNHIM il y a 7 ans, mais compte tenu des progrès impressionnants dans ce domaine, il n’était pas inutile de faire à nouveau une évaluation de la prise en charge thérapeutique actuelle de la polyarthrite rhumatoïde. Apres avoir traité dans une première partie les nouvelles biothérapies ciblant les cellules du système immunitaire, rituximab et abatacept (Cf. Dossier du CNHIM XXXI-4-2010), cette seconde partie (Dossier du CNHIM XXXI-5-2010) analyse en détail les modalités d’utilisation et les résultats cliniques de trois nouvelles biothérapies ciblant les cytokines, tocilizumab, certolizumab, golimumab.
Docteur Hubert Marotte
Service de rhumatologie CHU Saint-Etienne
Saint-Priest-en-Jarez
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies ciblant les cytokines :
tocilizumab, certolizumab, golimumab
Editorial

-6-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
La polyarthrite rhumatoïde (PR) est le plus fréquent des rhumatismes inflammatoires chroniques ; c’est une pathologie auto-immune. Une première partie de cet article dédié à la prise en charge de cette maladie, parue dans Dossier du CNHIM 2010–XXXI-4, traitait de l’épidémiologie de la PR, de sa physiopathologie, de son diagnostic, et en ce qui concerne sa prise en charge thérapeutique, des traitements symptomatiques, des traitements de fond (avec les critères d’évaluation de leur efficacité), ainsi que des nouvelles biothérapies ciblant les cellules du système immunitaire, le rituximab et l’abatacept. Cette deuxième partie traite des nouvelles biothérapies dont la cible thérapeutique est la cytokine IL-6 (tocilizumab) ainsi que deux nouveaux anti-TNFα (certolizumab pegol et golimumab). Ces nouvelles biothérapies enrichissent l’arsenal thérapeutique de la PR dont il convient d’optimiser l’utilisation afin d’induire une rémission pour le maximum de malades. Le tocilizumab est un anticorps monoclonal recombinant de type IgG1, dirigé contre les récepteurs de l’IL-6. L’IL-6 est une cytokine pro-inflammatoire impliquée dans la pathogenèse de la PR ; les malades atteints de PR présentent des concentrations élevées d’IL-6 et de récepteurs solubles de l’IL-6 dans le liquide synovial. L’efficacité du tocilizumab sur l’amélioration des signes cliniques et des symptômes de la PR a été évaluée au cours de plusieurs études cliniques. Il est indiqué dans le traitement de la PR active, modérée à sévère, chez les malades adultes, en association au méthotrexate (MTX) en cas de réponse inadéquate ou d’intolérance à au moins un traitement de fond ou en monothérapie en cas d’intolérance au MTX.
La posologie est de 8 mg/kg en perfusion IV toutes les 4 semaines. Un plan de gestion des risques (PGR) a été mis en place au niveau européen avec une surveillance spécifique des effets indésirables comme les infections graves, et la tenue de registres évaluant le profil de sécurité à long terme du tocilizumab. Le certolizumab est un fragment Fab’ d’anticorps humanisé recombinant, dirigé contre le TNF-α. Le certolizumab se lie au TNF-α avec une grande affinité et le neutralise. L’efficacité du certolizumab pegol administré par voie sous-cutanée a été évaluée dans plusieurs études cliniques. Il est indiqué dans le traitement de la PR active, modérée à sévère, chez les malades adultes en association au MTX, en cas de réponse inadéquate aux traitements de fond ou en monothérapie en cas d’intolérance au MTX. La posologie est de 400 mg en dose initiale, suivi de 400 mg à S2 et S4 puis 200 mg toutes les 2 semaines. Un plan de gestion des risques a été mis en place pour surveiller les risques infectieux et carcinogènes. Le golimumab est un anticorps monoclonal humain de type IgG1 produit sur une lignée cellulaire d’hybridome de souris par ADN recombinant. L’efficacité clinique du golimumab a été évaluée par plusieurs études cliniques. Le golimumab est indiqué dans le traitement de la PR active modérée à sévère en association avec le MTX, en cas de réponse inadéquate aux traitements de fond. La posologie est de 50 mg en SC une fois par mois.
Mots clés : anticorps monoclonal, biothérapie, certolizumab, golimumab, interleukine, maladie auto-immune, polyarthrite rhumatoïde, rhumatisme, TNF-α, tocilizumab.
Abstract.
Rheumatoid arthritis (RA) is the most common chronic inflammatory arthritis; it is an autoimmune disease. The first part of this article about the management of this pathology, published in Dossier CNHIM 2010-XXXI(4), dealt with the epidemiology, pathophysiology, diagnosis, and its therapeutic management including symptomatic treatment, DMARDs (and evaluation criteria), and new biological therapies targeting immune system cells ie rituximab and abatacept. This second part deals with new biological agents whose therapeutic target is the cytokine IL-6 (tocilizumab) and two new anti-TNFα (certolizumab pegol and golimumab). These new biological therapies should optimize the management of PR in order to induce remission for the larger number of patients. Tocilizumab is a recombinant monoclonal IgG1 directed against the receptors for IL-6. IL-6 is a proinflammatory cytokine involved in the pathogenesis of RA; the RA patients have high levels of IL-6 and soluble receptors of IL-6 in synovial fluid. The efficacy of tocilizumab in improving clinical signs and symptoms of RA was assessed in several clinical studies. It is indicated for the treatment of active RA, moderate to severe in adult patients, in combination with methotrexate (MTX) in case of inadequate response or intolerance to at least one DMARD monotherapy or in case of intolerance to MTX. The dose is 8 mg/kg IV infusion every 4 weeks. A risk management
plan was established at European level with specific monitoring of side effects such as severe infections and registries evaluating the profile of long-term safety of tocilizumab. Certolizumab is a Fab' fragment of recombinant humanized antibody directed against TNF-α. Certolizumab binds to TNF-α with high affinity and neutralizes it. The efficacy of certolizumab pegol administered subcutaneously has been evaluated in several clinical studies. Certolizumab is indicated for the treatment of active RA, moderate to severe in adult patients, in combination with MTX inadequate response to DMARDs or as monotherapy in case of intolerance to MTX. The initial dose is 400 mg followed by 400 mg on W2 and on W4, then 200 mg every 2 weeks. A risk management plan was put in place to monitor risks of infection and carcinogens. Golimumab is a human monoclonal IgG1 produced on a hybridoma cell line of mice by recombinant DNA. The clinical evaluation of golimumab was evaluated by several clinical studies. Golimumab is indicated for the treatment of moderate to severe active RA in combination with MTX in case of inadequate response to DMARDs. The dosage is 50 mg SC once a month.
Key words: abatacept, autoimmune disease, biotherapy, immunosuppressor, interleukin, monoclonal antibody, rheumatoid arthritis, rheumatism, rituximab, TNF-α.
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies ciblant les cytokines :
tocilizumab, golimumab, certolizumab
En bref Marie-Caroline HUSSON
Rédactrice en chef

-7-
Dossier du CNHIM, 2010, XXXI, 4
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Cet article contient des acronymes explicités en p. 56.
1. Introduction Cette deuxième partie de l’article sur la Polyarthrite Rhumatoïde (PR) et les nouvelles biothérapies, fait suite au Dossier du CNHIM 2010, XXXI-4 qui traitait de :
- l’épidémiologie de la PR, - sa physiopathologie, - son diagnostic, - sa prise en charge thérapeutique limitée aux traitements sympto-matiques, traitements de fond (et leurs critères d’évaluation), et les nouvelles biothérapies ciblant les cellules du système immunitaire, le rituximab et l’abatacept.
Dans cette seconde partie, d’autres nouvelles biothérapies seront traitées : celle dont la cible théra-peutique est le récepteur de la cytokine IL-6, le tocilizumab, ainsi que celles dont la cible est le TNF-α, le certolizumab pegol et le golimumab.
Ces nouvelles biothérapies enri-chissent l’arsenal thérapeutique de la PR dont il convient d’optimiser l’utilisation afin d’atteindre, pour le maximum de malades, les objectifs essentiels suivants : une efficacité clinique ou rémission (maîtrise de l’activité de la maladie), l’amélioration des capacités fonctionnelles (qualité de vie), la réduction des signes généraux (dont la diminution de la fatigue, diminution de la CRP), et la prévention et le contrôle des dommages structuraux.
La polyarthrite rhumatoïde (PR) est le plus fréquent des rhumatismes inflammatoires chroniques ; c’est une pathologie auto-immune. Elle se caractérise par une atteinte inflammatoire de la membrane synoviale des articulations et des tendons et se manifeste par des douleurs et des gonflements articulaires. La vitesse d’installation de la maladie est très variable d’un malade à l’autre.
Cette pathologie évolue par poussées et selon des schémas de gravité hétérogènes d’un malade à l’autre (formes faibles à modérées, formes agressives). Des manifestations extra-articulaires sont susceptibles de l’aggraver et d’engager le pronostic vital.
En l’absence de traitement, la PR entraîne des destructions articulaires et des déformations. Un handicap fonctionnel et une altération de la qualité de vie s’installent souvent de façon progressive. Pour le malade les répercussions sont multiples, à la fois fonctionnelles, psychologiques, sociales et professionnelles.
La prise en charge théra-peutique de la PR repose sur : - des traitements symptomatiques, sans action sur la prévention des lésions structurales, - et des traitements de fond, capables de freiner l’évolution des lésions structurales, désignés par la terminologie anglo-saxonne, Disease Modifying AntiRheumatic Drug (DMARD).
A côté, et aussi en complément des traitements de fond classiques comme le méthotrexate, d’autres traitements depuis une dizaine d’années ont modifié la prise en charge de la PR. Ces derniers, appelés biothérapies, ont des cibles thérapeutiques bien différenciées. Les premières biothérapies visaient deux cytokines pro-inflammatoires, le Tumor Necrosis Factor alpha (TNFα) et l’IL-1β.
Plus récemment, les nouvelles thérapeutiques développées visent : - des cellules du système immunitaire, les lymphocytes T (LT),
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies ciblant les cytokines : tocilizumab, golimumab, certolizumab
Emmanuelle RADIDEAU1, Salmane BAH1, Christine DUPONT1, Pascal HILLIQUIN2, et la participation du comité de rédaction
1 Service de pharmacie, CH Sud Francilien, Corbeil Essonnes. 2 Service de rhumatologie, CH Sud Francilien, Corbeil Essonnes.
Remerciements : Aurore Gouraud (Lyon), Hubert Marotte (Saint-Priest-en-Jarez), Alain Saraux (Brest), Elisabeth Solau (Poitiers).
En bref
La polyarthrite rhumatoïde (PR) est le plus fréquent des rhuma-tismes inflammatoires chroni-ques ; c’est une pathologie auto-immune. Une première partie de cet article dédié à la prise en charge de cette maladie, parue dans le Dossier du CNHIM 2010–XXXI-4, traitait de l’épidémiologie de la PR, de sa physiopathologie, de son diagnostic, et en ce qui concerne sa prise en charge thérapeutique, des traitements symptomatiques, de fond (avec les critères d’évaluation de leur efficacité), ainsi que des nouvelles biothérapies ciblant les cellules du système immunitaire, rituximab et abatacept. Cette deuxième partie traite des nouvelles bio-thérapies dont la cible thérapeutique est la cytokine IL-6 (tocilizumab) ainsi que deux nouveaux anti-TNFα (certolizu-mab pegol et golimumab). Ces nouvelles biothérapies enrichis-sent l’arsenal thérapeutique de la PR dont il convient d’optimiser l’utilisation afin d’induire une rémission pour le maximum de malades.

-8-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
avec l’abatacept, ou les lymphocytes B (LB), avec le rituximab ; - d’autres cytokines, comme l’interleukine 6 (IL-6), avec le tocilizumab.
La réponse thérapeutique peut être évaluée par différents critères.
Les critères de l’American College of Rheumatology (ACR) permettent d’évaluer la réponse d’un malade à son traitement. Un malade est qualifié de « répondeur » selon l’ACR 20 si sont obtenues : - une diminution d’au moins 20 % à la fois du nombre d’articulations douloureuses (NAD) et du nombre d’articulations tuméfiées (ou synovites ou NAG), et - une amélioration d’au moins 20 % du score sur 3 des 5 critères suivants : . évaluation de la douleur par le malade, . évaluation globale de l’activité de la PR par le médecin, . évaluation par le malade de son handicap fonctionnel, . taux de protéine C active, . vitesse de sédimentation. L’ACR 20 constitue le seuil minimum considéré à obtenir pour un effet thérapeutique recherché. Dans la plupart des études cliniques, le seuil de 20 % a été retenu comme critère principal de jugement. Des critères plus exigeants sont visés avec la réponse selon l’ACR 50 ou l’ACR 70.
La réponse thérapeutique EULAR sur l’activité de la maladie est jugée d’après : . l’amplitude de la diminution du critère « Disease Activity Score » ou de son amélioration, dans sa version DAS 28 (indice composite d’activité de la PR développé sur les articulations, excepté celles des hanches, avant-pieds et chevilles), . le niveau de l’activité de la maladie, soit la nouvelle valeur obtenue au DAS 28 Elle classe les malades traités en trois catégories : les non-répondeurs, les répondeurs modérés et les bons répondeurs. Le contrôle de l’activité de la maladie s’obtient avec un DAS 28 inférieur à 3,2, une rémission avec un DAS 28 inférieur à 2,6. Une amélioration du DAS 28 supérieure ou égale à 1,2 sous l’effet d’un traitement est significative.
La périodicité de l’évaluation de l’activité de la maladie est fonction du contexte notamment, si la PR est contrôlée ou non. Cette évaluation repose à la fois sur la clinique et sur la biologie (Vitesse de sédimentation (VS), taux de protéine C activée (CRP).
Le délai de 3 mois est retenu, en particulier au début de la maladie. Dans le cas d’une PR très évolutive, il peut être raccourci à 1 mois. A l’inverse, lorsque la PR est en rémission, ce délai peut être allongé à 6 mois.
La mesure de la qualité de vie fonctionnelle peut être réalisée par l’outil Health Assessment Question-naire Disability Index (HAQ-DI). Il cherche ainsi à évaluer certaines activités journalières en relation avec l’incapacité fonctionnelle spéci-fique de la PR.
L’évaluation de la fatigue ressentie par le malade dans les 7 jours précédents peut être faite par un auto-questionnaire malade composé de 13 questions, cotées de 0 à 4, couvrant 4 domaines. Le score global peut varier de 0 (fatigue extrême) à 52 (absence de fatigue). Plus le score est haut, moins la fatigue est importante. Une augmentation supérieure ou égale à 4 points est généralement jugée cliniquement pertinente. FACIT - Fatigue ou Functional Assessment of Chronic Illness Therapy.
Les radiographies simples suffisent pour le suivi. Mais l’analyse peut être standardisée à l’aide d’indices fiables et validés tels que l’indice de Sharp. Ces indices cotent la sévérité de l’atteinte radiologique de chaque articulation. Ils sont utilisés dans les études cliniques, leur réalisation peut être longue. Le score de Sharp étudie séparément en 2 sous-scores, les érosions et le pincement de l’interligne articulaire. Le score d’érosion de Sharp modifié par Genant évalue les articulations des deux mains.
En fonction des AMM (HAS), les traitements s’adressent à une : - PR active : en fonction de la valeur du DAS 28, - PR sévère : elle se définit par l’existence d’un handicap fonctionnel mesuré par l’HAQ (supérieur ou égal à 0,5), ou par l’existence ou la
progression de lésions structurales en imagerie, ou par l’existence de manifestations systémiques. Un seul critère suffit. - PR évolutive : elle se définit comme une PR active ou avec une progression structurale ou fonction-nelle dans le temps. - PR grave : il s’agit d’une PR sévère ou justifiant pour son contrôle un traitement de fond continu.
2. Tocilizumab
2.1. Renseignements gé-néraux et galéniques
Cf. tableau I
2.2. Propriétés pharmaco-logiques
2.2.1. Mode d’action
L’interleukine-6 (IL-6) (1), cytokine pro-inflammatoire, est impliquée dans la pathogénie de la PR, tant au niveau local que systémique.
En bref Le tocilizumab est un anticorps monoclonal recombinant de type IgG1, dirigé contre les récepteurs de l’IL-6. L’IL-6 est une cytokine pro-inflammatoire impliquée dans la pathogenèse de la PR ; les malades atteints de PR présentent des concentrations élevées d’IL-6 et de récepteurs solubles de l’IL-6 dans le liquide synovial. L’efficacité du tocilizumab sur l’amélioration des signes clini-ques et des symptômes de la PR a été évaluée au cours de plusieurs études cliniques. Il est indiqué dans le traitement de la PR active, modérée à sévère, chez les malades adultes, en association au méthotrexate (MTX) en cas de réponse inadé-quate ou d’intolérance à au moins un traitement de fond ou en monothérapie en cas d’into-lérance au MTX. La posologie est de 8 mg/kg en perfusion IV toutes les 4 semaines. Un plan de gestion de risque (PGR) a été mis en place au niveau européen avec une surveillance spécifique des effets indésirables comme les infections graves, et des registres évaluant le profil de sécurité à long terme du tocilizumab.

-9-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau I. Renseignements généraux et galéniques (42, 45)
DCI, Spécialité, Laboratoire
Forme galénique Dosage
Présentation
Excipients AMM, Liste,
Remboursement Conservation
Tocilizumab ROACTEMRA® Chugaï Roche
Solution à diluer pour perfusion 20 mg/ml. Boîte de 1 flacon. Flacon de 4 ml contient 80 mg. Flacon de 10 ml contient 200 mg. Flacon de 20 ml contient 400 mg.
Saccharose, polysorbate 80,
phosphate disodique dodécahydraté,
phosphate monosodique dihydraté,
eau PPI
. AMM européenne 2009 Liste I, Collectivités. . Inscription sur la liste des spécialités pharmaceutiques prises en charge en sus des prestations d’hospitalisation selon l’Arrêté du 9/08/87. . Médicament réservé à l’usage hospitalier. . Prescription restreinte réservée aux spécialistes en rhumatologie ou en médecine interne. . Surveillance particulière pendant le traitement.
. Conservation entre +2°C et +8°C . Conserver les flacons dans l’emballage extérieur à l’abri de la lumière. . Ne pas congeler
Son action est médiée par deux types de récepteurs, membranaires et solu-bles.
L’IL-6 intervient ainsi dans le processus inflammatoire aigu (local et systémique) et dans l’inflammation chronique :
. elle induit la synthèse hépatique des protéines de la phase aiguë (Protéine C réactive (CRP), protéine sérique amyloïde A, notamment) et stimule le recrutement local des polynucléaires neutrophiles ;
. elle joue un rôle dans le passage de l’inflammation aiguë à l’inflammation chronique ; elle participe ainsi au recrutement et à l’activation des macrophages qui sécrètent à leur tour des cytokines pro-inflam-matoires.
L’Il-6 joue aussi un rôle dans l’immunité humorale et l’immunité cellulaire : différenciation des lym-phocytes B en plasmocytes sécré-tants (sécrétion des immunoglo-bulines et notamment des anticorps antiprotéines citrullinés (anti-CCP) et du facteur rhumatoïde (FR)), différenciation et activation des lymphocytes T et en particulier Th17. Au niveau osseux, l’IL-6 favorise l’ostéoclastogenèse et participe donc à la destruction ostéo-articulaire. L’Il-6 intervient dans l’induction de la synthèse du VEGF (médiateur de
l’angiogenèse ou facteur de crois-sance de l’endothélium vasculaire) par les fibroblastes synoviaux.
Au plan systémique, l’IL-6 participe aussi au développement de certains symptômes fréquents chez le malade atteint de PR, tels que l’anémie et la fatigue.
Chez les malades atteints de PR, les concentrations de l’IL-6 et de ses récepteurs solubles (sIL-6R) dans le liquide synovial et au niveau sérique sont élevées. Les concentrations sériques d’IL-6 sont corrélées positivement avec l’activité de la PR.
Comme le montre la figure 1, l’IL-6 peut induire un signal de deux manières : - en se fixant directement sur son récepteur membranaire associé à la glycoprotéine gp 130, - en se fixant sur son récepteur soluble, et le complexe ainsi formé se fixe sur la glycoprotéine gp 130 transmembranaire.
Le tocilizumab est un anticorps monoclonal recombinant humanisé, de type Ig G1, dirigé contre les récepteurs de l’IL-6, solubles et membranaires. Il se lie spécifique-ment à ces 2 types de récepteurs, de façon compétitive (2) et : . il empêche la dimérisation de la glycoprotéine gp 130 à la surface des cellules,
. il bloque la transmission du signal de l’IL-6 au niveau cellulaire. In vivo, le maximum (> 90 %) de saturation des récepteurs solubles à l’IL-6 est atteint quand les concentrations de tocilizumab dans le sérum sont supérieures à 1 µg/mL (2).
Le signal de l’IL-6 est inhibé aussi longtemps que le tocilizumab libre est détecté au niveau sérique comme l’indique la normalisation des marqueurs de l’inflammation et le remodelage du cartilage et de l’os.
2.2.2. Pharmacocinétique
A l’état d’équilibre, après une perfusion de tocilizumab de 8 mg/kg toutes les 4 semaines pendant 24 semaines, les valeurs suivantes ont été obtenues (2) : . Pour l’AUC : 35,0 mg.h /mL, . Pour la Cmin : 9,7 µg/mL, . Pour la Cmax : 183 µg/mL.
L’état d’équilibre a été atteint après la première administration de tocilizumab pour la Cmax, et après 8 et 20 semaines respectivement pour l’AUC et la Cmin (2).
Dans le cas de l’administration de tocilizumab à la dose de 8 mg/kg au lieu de 4 mg/kg, la valeur de l’AUC est multipliée par 2,7 et celle de la Cmax par 6,5 (2).

-10-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Figure 1. Mécanisme d’action du tocilizumab (d’après Okuda Y., Biologics : Targets and Therapy, 2008) Distribution : Chez les malades atteints de PR, le volume central de distribution a été estimé à 3,5 L, le volume périphé-rique de distribution a été de 2,9 L, ce qui conduit à un volume de distribution à l’état d’équilibre de 6,4 L (la dose de tocilizumab administrée n’est pas précisée) (2).
Elimination : Après administration intraveineuse, le tocilizumab est éliminé de la circulation selon un mode biphasique. La clairance totale du tocilizumab est dépendante de la concentration, et égale à la somme des clairances linéaire et non linéaire. La valeur de la clairance linéaire a été estimée à 12,5 mL/h dans l’analyse pharmacocinétique de population. La clairance non linéaire, dépendante de la concentration, joue un rôle majeur aux concentrations faibles de tocilizumab.
Lorsque la voie de la clairance non linéaire est saturée, à des concentrations plus élevées de tocilizumab, la clairance est principalement déterminée par la clairance linéaire (2).
La demi-vie (T1/2) de tocilizumab est dépendante de la concentration.
A l’état d’équilibre, après une dose de 8 mg/kg administrée toutes les 4 semaines, la T1/2 effective a diminué de 14 à 8 jours au fur et à mesure que les concentrations diminuent entre deux perfusions. La clairance de tocilizumab n’est pas affectée par l’administration concomitante de MTX. L’âge, le sexe et l’origine ethnique n’affectent pas les paramètres pharmacocinétiques du tocilizumab (2).
2.3. Efficacité clinique
L’efficacité du tocilizumab sur l’amélioration des signes cliniques et des symptômes de la PR a été évaluée au cours de : . deux études japonaises, l’étude SAMURAI (cf. tableau II) (3) et l’étude SATORI (cf. tableau III) (4) ; . six études multicentriques, internationales, randomisées en double aveugle.
Pour ces dernières études, les malades présentaient soit :
. une réponse inadéquate ou une intolérance au MTX : études CHARISMA (cf. tableau IV) (5), OPTION (cf. tableau VI) (7) et LITHE (10) ;
. une réponse inadéquate ou une intolérance à au moins un DMARD : étude TOWARD (cf. tableau VII) (8) ;
. une réponse inadéquate ou une intolérance à un ou plusieurs anti-TNF : étude RADIATE (cf. tableau V) (6) ;
. une absence de traitement récent par méthotrexate : étude AMBITION (cf. tableau VIII) (9).
Ces études ont inclus des malades âgés d’au moins 18 ans atteints de PR active diagnostiquée selon les critères de l’ACR. A l’inclusion, ils présentaient au moins six articu-lations douloureuses et huit articulations gonflées.
Le critère de jugement principal de chacune de ces études a été la proportion de malades atteignant une réponse ACR 20 à la semaine 24.
Dans l’étude LITHE, le critère de jugement principal comprenait en plus l’évolution du score total de Sharp modifié par Genant et l’évolution de la fonction physique mesurée par le questionnaire HAQ à 104 semaines.
Récepteur soluble à l’IL-6

-11-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau II. Etude CHARISMA – étude de phase IIb randomisée, évaluant l’efficacité et la tolérance du tocilizumab, seul ou en association au méthotrexate dans la polyarthrite rhumatoïde active (5)
Etude CHARISMA. The Chugaï Humanized Anti Human Recombinant Interleukin-6 Monoclonal Antibody – 2006 (5).
Méthodologie Objectif Evaluer l’efficacité et la tolérance du tocilizumab seul ou associé au méthotrexate chez des malades atteints de PR active avec une réponse inadéquate au métho-trexate (MTX). Type d’étude Etude de phase IIb, européenne, randomisée en double aveugle. Schéma et posologie Les malades reçoivent leur dose habituelle de méthotrexate pendant une période de stabili-sation d’au moins 4 semaines, puis ils sont randomisés dans l’un des 7 bras de traitement suivants : BRAS 1 : Tocilizumab 2 mg/kg + placebo du méthotrexate BRAS 2 : Tocilizumab 4 mg/kg + placebo du méthotrexate BRAS 3 : Tocilizumab 8 mg/kg + placebo du méthotrexate BRAS 4 : Tocilizumab 2 mg/kg + méthotrexate BRAS 5 : Tocilizumab 4 mg/kg + méthotrexate BRAS 6 : Tocilizumab 8 mg/kg + méthotrexate BRAS 7 : placebo du tocilizumab + méthotrexate Administration du tocilizumab ou de son placebo toutes les 4 semaines. Méthotrexate : 10-25 mg/semaine Durée : 5 mois soit 20 semaines (période de stabilisation comprise).
Inclusion – Evaluation Inclusion . Malades atteints de PR≥ 6 mois. . Nombre d’articulations gonflées ≥ 6 et nombre d’articulations douloureuses ≥ 6. . CRP≥ 10 mg/L. . VS ≥ 28 mm/h. . Malades sous MTX (10-25 mg/semaine). . Corticoïdes : prednisolone ou équivalent < 10 mg/jour (dose stable depuis au moins 4 semaines). Exclusion . Leucopénie < 4 G/L. . Taux des PNN < 2 G/L. . Taux des Plaquettes < 150 G/L. . ASAT et ALAT > 1,5 N.
Evaluation en ITT Critère principal : ACR 20 à 16 semaines Critères secondaires : ACR 50 et ACR 70, paramètres individuels de l’ACR, critère HAQ, DAS 28, raideur matinale, VS, CRP.
Résultats Nombre de malades 359 malades randomisés : Bras 1 : 53 - Bras 2 : 54 - Bras 3 : 52 - Bras 4 : 52 - Bras 5 : 49 - Bras 6 : 50 - Bras 7 : 49. Analyse des malades L’analyse a porté sur 354 malades (5 : violation de protocole). 60 sortis de l’étude, dont 34 pour EI. Pour les EI, la répartition est la suivante : Bras 1 : 4 – Bras 2 : 6 – Bras 3 : 5 – Bras 4 : 3 – Bras 5 : 6 – Bras 6 : 6 – Bras 7 : 4. Pourcentage de malades ayant complété l’étude (4 mois) : Bras 1 : 77 % (41 malades) - Bras 2 : 79 % (43) - Bras 3 : 84 % (44)- Bras 4 : 88 % (46) - Bras 5 : 86 % (42) - Bras 6 : 86 % (43) - Bras 7 : 82 % (40).
Profil des malades Les 7 groupes sont comparables. Age moyen : 50,3 ans. Pourcentage de femmes : 78,5 %. Ancienneté de la maladie : 9 ans (de 7,82 (bras 5) à 11,24 (bras 7). CRP (mg/L) : 27. DAS 28 : 6,5. Durée de traitement sous méthotrexate : de 29 à 40,4 mois selon les groupes. Résultats cliniques La monothérapie de tocilizumab à la dose de 4 mg/kg ou 8 mg/kg, ainsi que toutes les associations ont montré un résultat significatif par rapport à l’association placebo + méthotrexate (p<0,05) sur les critères ACR 20. Seule la combinaison tocilizumab 8 mg/kg + méthotrexate a montré un résultat significatif par rapport à l’association placebo + métho-trexate (p<0,05) sur les critères ACR 50 et 70. …/…

-12-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau II (suite et fin). Etude CHARISMA – étude de phase IIb randomisée, évaluant l’efficacité et la tolérance du tocilizumab, seul ou en association au méthotrexate dans la polyarthrite rhumatoïde active (5)
Résultats cliniques (suite)
Réponses ACR à S16
Tocilizumab 2 mg/kg + placebo
Tocilizumab 4 mg/kg + placebo
Tocilizumab 8 mg /kg + placebo
Placebo du tocilizumab + MTX
ACR 20 31 % 61 % (p < 0,05) 63 % (p < 0,05) 41 %
ACR 50 6 % 28 % 41 % 29 % ACR 70 2 % 6 % 16 % 16 %
Réponses ACR à S16
Tocilizumab 2 mg/kg + MTX
Tocilizumab 4 mg/kg + MTX
Tocilizumab 8 mg/kg + MTX
Placebo du tocilizumab + MTX
ACR 20 64 % (p < 0,05) 63 % (p< 0,05) 74 % (p = 0,001) 41 %
ACR 50 32 % 37 % 53 % (p < 0,05) 29 % ACR 70 14 % 12 % 37 % (p < 0,05) 16 %
DAS 28 A l’exception du groupe recevant le tocilizumab 2 mg /kg en monothérapie, tous les groupes recevant le tocilizumab seul ou associé ont montré une diminution significative du DAS 28 à S 16 par rapport au groupe témoin, placebo + méthotrexate : pour la combinaison et la monothérapie avec tocilizumab 8 mg/kg : p<0,001 ; pour la combinaison à 2 et 4 mg/kg : p< 0,05 ; pour la monothérapie tocilizumab à 4 mg/kg : p<0,05. Le taux de malades en rémission a été de 34 % dans le groupe tocilizumab 8 mg/kg + méthotrexate contre 17 % dans le groupe tocilizumab 8 mg/kg en monothérapie et 8 % dans le groupe méthotrexate + placebo.
Composants individuels de la réponse ACR Tous les composants individuels de la réponse clinique (nombre d’articulations douloureuses, d’articulations gonflées, durée de la raideur matinale, HAQ, évaluation de la douleur par EVA patient, et évaluations globales de la maladie par le médecin et le malade) ont été améliorées par des doses de tocilizumab supérieures à 4 mg/kg. La diminution moyenne du nombre d’articulations gonflées observée avec la combinaison à 8 mg/kg et la monothérapie est significativement plus importante par rapport à celle observée sous placebo plus méthotrexate (p = 0,01 et p < 0,01). Un résultat identique est observé pour la diminution du nombre d’articulations douloureuses (p=0,009) Effets indésirables - Infections sévères : 4 cas dans le bras 1 et 3 cas dans le bras 6. Absence de cas de tuberculose. - Réactions d’hypersensibilité : 4 cas dans le bras 1 et 2 cas dans le bras 2, 1 dans le bras 4. - Elévation modérée. et transitoire des transaminases chez environ 40 % des malades traités par tocilizumab. L’augmentation maximale des enzymes ALAT/ASAT à la semaine 2 a été de 45 % dans le bras 3 et de 88 % dans le bras 6 - Elévation modérée et transitoire de la bilirubine chez certains malades recevant du tocilizumab. Parmi les malades sous tocilizumab en monothérapie, l’augmentation maximale de la bilirubine a été atteinte à la semaine 16 (83 %). Parmi les malades recevant la combinaison tocilizumab plus méthotrexate, le maximum d’augmentation de la bilirubine a été observé à la semaine 14 (59 % dans le bras 5). - Diminution des neutrophiles réversible et dose dépendante sous tocilizumab. 13 cas de neutropénie grade 2 ou 3 dans les groupes traités par 8 mg/kg de tocilizumab. A la semaine 14, la réduction des polynucléaires neutrophiles a été de 47 % dans le bras 3 et de 43 % dans le bras 6. - Présence d’anticorps anti-tocilizumab détectée chez 25 malades appartenant exclusivement aux bras 1 et 2 (tocilizumab en monothérapie).
Conclusion des auteurs
Les résultats de cette étude montrent l’efficacité de l’inhibition de l’IL-6 sur l’activité de la PR. La tolérance du traitement par le tocilizumab n’est pas modifiée par l’association au méthotrexate.
Conclusion du CNHIM
Les associations de tocilizumab (quelle que soit la dose) et de méthotrexate, et les monothérapies de tocilizumab (à partir de 4 mg/kg) ont conduit à une amélioration significative clinique selon le critère ACR 20 versus placebo + méthotrexate (p<0,05). Le degré de significativité a été plus important pour l’association tocilizumab 8 mg/kg plus méthotrexate (p=0,001). Seule l’association tocilizumab 8 mg/kg plus méthotrexate améliore les signes cliniques selon l’ACR 50 ou 70. Cette étude qui s’apparente à une étude de doses a porté sur des effectifs corrects mais la durée d’évaluation a été courte (inférieure à 6 mois).

-13-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau III. Etude SAMURAI – étude japonaise de phase III, randomisée, évaluant l’efficacité et la tolérance du tocilizumab en monothérapie dans la polyarthrite rhumatoïde (3)
Etude SAMURAI. Study of Active controlled Monotherapy Used for Rheumatoid Arthritis, an IL-6 Inhibitor trial – 2007 (3).
Méthodologie
Objectif Evaluer l’efficacité et la tolérance du tocilizumab en monothérapie sur l’inhibition de la progression de la destruction structurale des articulations des malades atteints de PR. Type d’étude Etude de phase III, Japonaise (sur 28 sites), randomisée. Schéma et posologie 2 bras de traitement : BRAS 1 : DMARDs conventionnels : la dose, le type et l’association de DMARD et/ou immunosuppresseur (à l’exception des anti-TNF et du léflunomide) déterminés selon l’activité de la PR et à la discrétion de l’investigateur. BRAS 2 : Tocilizumab en monothérapie : une perfusion de 8 mg/kg toutes les 4 semaines. Durée : 52 semaines.
Inclusion - Evaluation
Inclusion . Malades âgés de plus de 20 ans atteints de PR ≥ 6 mois . PR diagnostiquée selon les critères ACR : articulations gonflées ≥ 6 et articulations douloureuses ≥ 6 ; une CRP ≥ 20 mg/L ; une VS ≥ 30 mm/h – une réponse inadéquate à au moins un DMARD ou immunosuppresseurs . Aucune administration d’anti-TNF ou de léflunomide autorisée dans les 3 derniers mois précédant la première dose. . Corticoïdes autorisés : predni-solone ou équivalent, à une dose < 10 mg/j, stable depuis deux semaines au minimum. Exclusion . Antécédent d’allergie sévère, comorbidités importantes. . Infections récurrentes dans les 4 dernières semaines. Evaluation en ITT Critère principal : critère radio-graphique à S 28 et S 52 (selon Score de Sharp modifié par Van der Heijde, main et avant-pied) ; évaluation radiologique en aveugle. Critères secondaires : ACR 20, 50 et 70, DAS 28.
Résultats
Nombre de malades 306 malades randomisés : Bras 1 (DMARD) : 148 Bras 2 (tocilizumab) : 158 Analyse des malades . L’analyse a porté sur 300 malades : 143 dans le bras 1, 157 dans le bras 2. Quatre malades n’ont pas été traités. Aucune information n’est donnée sur 2 malades du groupe DMARD. . Malades arrivant au bout de l’étude (52 semaines) Bras 1 : 131 (90 %) Bras 2 : 134 (85 %) . Causes de sortie d’étude : Bras 1 : EI : 5, exacerbation de la maladie : 3, refus de traitement : 4, violation de protocole : 2 Bras 2 : EI : 17, exacerbation de la maladie : 1, refus de traite-ment : 1, violation de protocole : 1, AC anti-tocilizumab : 3 Profil des malades Les 2 groupes sont comparables. Age moyen: 53 ans Pourcentage de femmes : 81 % Ancienneté de la maladie (années) : 2,3 ans Nb de DMARDs antérieurs : 2,7 CRP (mg/L) : 48 DAS 28 : 6,4 Dose moyenne (mg) de MTX/sem. : 7 mg . 67 % des malades dans le groupe DMARDs reçoivent du méthotrexate : 37 % en association avec un ou plusieurs autres DMARDs ; 30 % seul . 22 % des malades reçoivent des DMARDs et/ou un immunosuppresseur autre que le méthotrexate. A l’initiation : TSS modifié (0-448) : environ 30 (score érosion : 13,8 ; score pincement : 15,4).
…/…
-14-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau III (suite et fin). Etude SAMURAI – étude japonaise de phase III, randomisée, évaluant l’efficacité et la tolérance du tocilizumab en monothérapie dans la polyarthrite rhumatoïde (3)
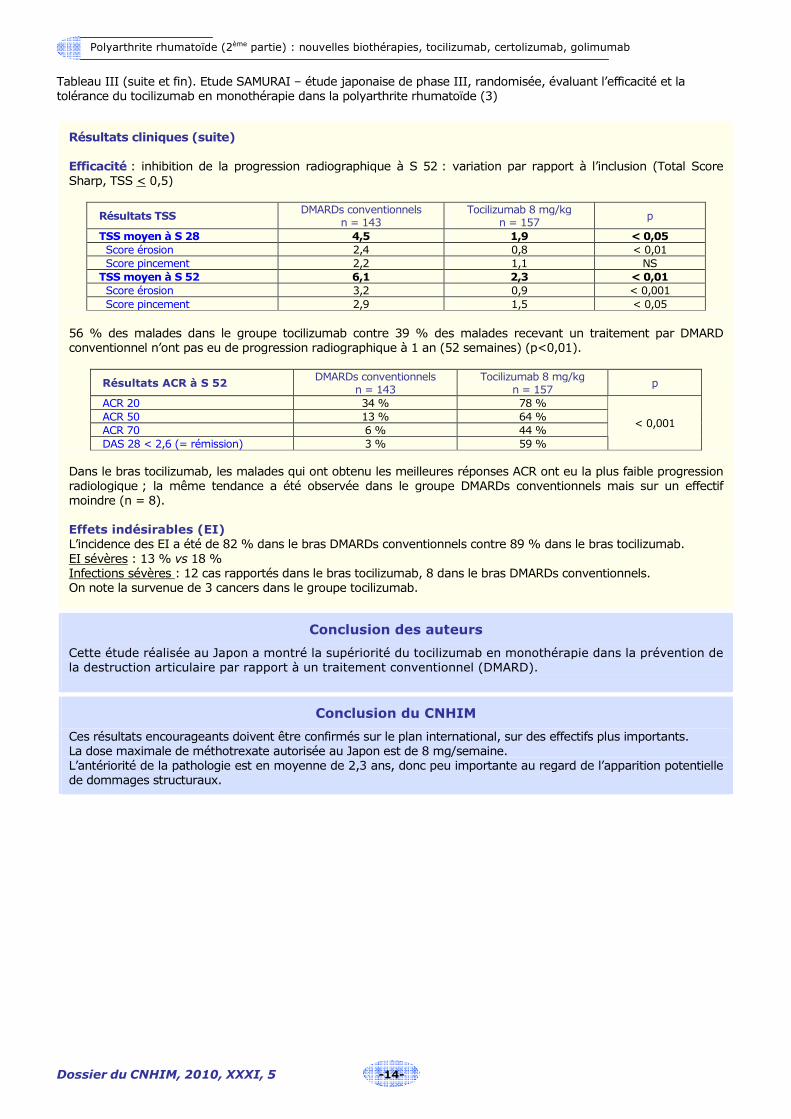
Résultats cliniques (suite) Efficacité : inhibition de la progression radiographique à S 52 : variation par rapport à l’inclusion (Total Score Sharp, TSS < 0,5)
Résultats TSS DMARDs conventionnels
n = 143 Tocilizumab 8 mg/kg
n = 157 p
TSS moyen à S 28 4,5 1,9 < 0,05 Score érosion 2,4 0,8 < 0,01 Score pincement 2,2 1,1 NS TSS moyen à S 52 6,1 2,3 < 0,01 Score érosion 3,2 0,9 < 0,001
Score pincement 2,9 1,5 < 0,05
56 % des malades dans le groupe tocilizumab contre 39 % des malades recevant un traitement par DMARD conventionnel n’ont pas eu de progression radiographique à 1 an (52 semaines) (p<0,01).
Résultats ACR à S 52 DMARDs conventionnels
n = 143 Tocilizumab 8 mg/kg
n = 157 p
ACR 20 34 % 78 % ACR 50 13 % 64 % ACR 70 6 % 44 % DAS 28 < 2,6 (= rémission) 3 % 59 %
< 0,001
Dans le bras tocilizumab, les malades qui ont obtenu les meilleures réponses ACR ont eu la plus faible progression radiologique ; la même tendance a été observée dans le groupe DMARDs conventionnels mais sur un effectif moindre (n = 8). Effets indésirables (EI) L’incidence des EI a été de 82 % dans le bras DMARDs conventionnels contre 89 % dans le bras tocilizumab. EI sévères : 13 % vs 18 % Infections sévères : 12 cas rapportés dans le bras tocilizumab, 8 dans le bras DMARDs conventionnels. On note la survenue de 3 cancers dans le groupe tocilizumab.
Conclusion des auteurs
Cette étude réalisée au Japon a montré la supériorité du tocilizumab en monothérapie dans la prévention de la destruction articulaire par rapport à un traitement conventionnel (DMARD).
Conclusion du CNHIM
Ces résultats encourageants doivent être confirmés sur le plan international, sur des effectifs plus importants. La dose maximale de méthotrexate autorisée au Japon est de 8 mg/semaine. L’antériorité de la pathologie est en moyenne de 2,3 ans, donc peu importante au regard de l’apparition potentielle de dommages structuraux.

-15-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau IV. Etude SATORI – étude japonaise de phase III randomisée, évaluant l’efficacité et la tolérance du tocilizumab en monothérapie dans la polyarthrite rhumatoïde présentant une réponse inadéquate au méthotrexate à faible dose (4)
Etude SATORI. Study of Active controlled TOcilizumab monotherapy for Rheumatoid arthritis patients with an Inadequate response to methotrexate – 2009 (4).
Méthodologie
Objectif Evaluer l’efficacité et la tolérance du tocilizumab en monothérapie chez les malades atteints d’une PR présentant une réponse inadéquate au méthotrexate à faible dose. Type d’étude Etude de phase III, randomisée, contrôlée, en double aveugle, Japonaise (25 sites). Schéma et posologie 2 bras de traitement : BRAS 1 : Tocilizumab 8 mg/kg toutes les 4 semaines, associé au placebo de méthotrexate une fois par semaine. BRAS 2 (groupe contrôle) : Placebo de tocilizumab toutes les 4 semaines, associé au méthotrexate 8 mg / semaine. Association à la prednisolone 10 mg/ jour au maximum, la dose ne pouvant pas être augmentée pendant l’étude. Durée : 24 semaines * VGEF = Facteur de croissance de l’endothélium vasculaire
Inclusion – Evaluation
Inclusion . Malades âgés de 20 à 75 ans atteints de PR ≥ 6 mois selon les critères ACR, avec : - un nombre d’articulations gonflées ≥ 6, - et un nombre d’articulations douloureuses ≥ 6 ; CRP ≥ 10 mg/L ou VS > 30 mm/h. . Malades sous méthotrexate 8 mg/sem. (dose maximale autorisée au Japon) durant les 8 dernières semaines avant la première injection. . Réponse inadéquate au MTX. . Absence : - de traitement anti-TNFα ou de léflunomide pendant les 12 semaines précédant la 1ère dose ; - d’échange plasmatique ou chirurgie pendant les 4 semaines précédant la 1ère dose, - de traitement par des DMARDs autre que le MTX ou par des immunosuppresseurs pendant les 2 semaines précédant la 1ère dose. . Taux de : Leucocytes > 3,5 G/L, Lymphocytes > 0,5 G/L.
Exclusion . PR au stade classe fonctionnelle IV ; . ASAT et ALAT > 1,5 N ; . Fibrose pulmonaire ou maladie pulmonaire active, épanchement pleural, ascite ; . Ag HBs et/ou Ac anti HCV positif(s) ; . Antécédents d’effet indésirable sévère au MTX ; . Pathologie associée cardiaque, hématologique, respiratoire, neurologique, endocrinienne, rénale, hépatique ou digestive significative ; . Infection nécessitant un traitement pendant les 4 semaines précédant la 1ère dose ; . Consommation excessive d’alcool ; . Réaction allergique sévère.
Evaluation Critère principal : ACR 20 à 24 semaines Critères secondaires : ACR 50 et ACR 70 à 24 semaines, DAS 28, HAQ, taux sérique de VEGF (*).
Résultats
Nombre de malades 127 malades randomisés : Bras 1 : 61 - Bras 2 : 66. Analyse des malades . Deux malades du bras 2 n’ont pas été traités. L’analyse a porté sur 61 malades du bras 1, 64 du bras 2. . Malades arrivant au bout de l’étude (S 24) : Bras 1 : 54 (89 %) Bras 2 : 33 (52 %) . Les causes de sortie de l’étude : Bras 1 : réponse inadéquate : 1, défaut d’observance : 2, effets secondaires : 2, violation de protocole : 2 Bras 2 : réponse inadéquate : 20, défaut d’observance : 4, effets secondaires : 3, à la demande des malades : 3, violation de protocole : 1 Profil des malades Les 2 groupes sont comparables Age moyen: 51,7 ans Pourcentage de femmes : 75 % (bras 2) à 90 % (bras 1) Ancienneté de la maladie : 8,6 ans Nombre de DMARDs antérieurs : 3,5 CRP (mg/L) : 31 DAS 28 : 6,1 VGEF : 721,05 pg/mL
…/…

-16-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau IV (suite et fin). Etude SATORI – étude japonaise de phase III randomisée, évaluant l’efficacité et la tolérance du tocilizumab en monothérapie dans la polyarthrite rhumatoïde présentant une réponse inadéquate au méthotrexate à faible dose (4).
Résultats cliniques (suite) Efficacité
A S 24 Bras 1 (tocilizumab) Bras 2 (placebo + MTX) p ACR 20 80,3 % 25 % < 0,001
ACR 50* 49,2 % 10,9 % < 0,001 ACR 70* 29,5 % 6,3 % < 0,001 DAS 28 score < 2,6 (rémission)* 43,1 % 1,6 % < 0,001
* dernière observation
. Critère HAQ : 67 % (bras 1) vs 34 % (p<0,001) . VGEF : La diminution de la concentration sérique du VGEF par rapport à la valeur initiale de base est plus importante dans le groupe tocilizumab que dans le groupe méthotrexate : à S 24, - 346,9 pg/mL bras 1 contre – 74,0 pg/mL bras 2 (p< 0,001). Effets indésirables (EI) 211 EI observés chez 56 des 61 malades (91,8 %) du bras 1 ; 104 EI chez 46 des 64 malades (71,9 %) du bras 2. E.I graves : 6,6 % (bras 1) vs 4,7 % (bras 2). Aucun cas de tuberculose n’a été observé. EI observés chez au moins 5 % des malades : - Hyperlipidémie 6,6% (bras 1) vs 1,6% (bras 2) - Diarrhées : 6,6% vs 1,6% - Rash : 6,6% vs 3,1% - Maux de tête : 6,6% vs 3,1% - Inflammation des voies respiratoires : 4,9 % vs 6,3% - Stomatites : 11,5 % vs 0 - Rhinopharyngite : 18 % vs 10,9%. Des réactions à l’injection ont été observées pour 7 malades (un total de 8 évènements) dans le groupe tocilizumab : prurit, maux de tête, flush, rash, arthralgie, augmentation tensionnelle).
Conclusion des auteurs
Cette étude a montré l’efficacité du tocilizumab, anti-récepteur IL-6, avec un rapport bénéfice/risque favorable.
Conclusion du CNHIM
Cette étude réalisée au Japon met en évidence des résultats cliniques en faveur du tocilizumab. Le nombre de malades et la durée de l’étude sont trop faibles pour évaluer correctement la sécurité du médicament. L’ancienneté moyenne de la pathologie est de 8,6 ans, les prises en charge sont donc plus anciennes que dans l’étude SAMURAI.

-17-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau V. Etude RADIATE – étude de phase III randomisée, évaluant l’efficacité et la tolérance du tocilizumab dans la polyarthrite rhumatoïde présentant une réponse inadéquate aux anti-TNF-α (6)
Etude RADIATE. The Research on Actemra Determining effIcacy after Anti-TNF failures – 2008 (6).
Méthodologie
Objectif Evaluer l’efficacité et la tolérance du tocilizumab chez des malades atteints d’une PR et présentant une réponse inadéquate ou une intolérance aux anti-TNF-α. Type d’étude Etude de phase III randomisée, contrôlée, en double aveugle contre placebo, multicentrique (Amérique du Nord, Europe de l’Ouest) Schéma et posologie 3 bras de traitement : BRAS 1 : Tocilizumab 8 mg/kg + méthotrexate BRAS 2 : Tocilizumab 4 mg/kg + méthotrexate BRAS 3 (contrôle) : Placebo + méthotrexate Méthotrexate : 10 à 25 mg/ semaine A S16, les malades non répondeurs pouvaient recevoir une association de tocilizumab 8 mg/kg + méthotrexate.
Durée : 24 semaines
Inclusion – Evaluation
Inclusion - Malades atteints de PR active, modérée à sévère depuis au moins 6 mois, avec un nombre d’articulations gonflées > 6 et un nombre d’articulations douloureu-ses ≥ 8 ; CRP ≥ 10 mg/L ; VS > 28 mm/h. - Malades intolérants ou réfrac-taires aux anti- TNFα. - Les traitements antérieurs ont été arrêtés : depuis au moins 2 semaines pour l’etanercept, depuis au moins 8 semaines pour l’infliximab et l’adalimumab, depuis au moins 12 semaines pour le léflunomide. Les DMARDS autre que le méthotrexate sont arrêtés avant l’inclusion. - Les malades devaient être traités par méthotrexate durant les 12 semaines précédant l’inclusion, avec une dose stable depuis au moins 8 semaines. Exclusion . Antécédent d’autre pathologie inflammatoire ou PR au stade IV fonctionnel. . Infections récurrentes ou antécédent néoplasique. . Immunodéficience primaire ou secondaire. . Taux d’hémoglobine (Hb) ≤ 8 g/dL. . Leucopénie, neutropénie, thrombopénie. . Fonction hépatique anormale. . Triglycérides (TG) > 10 mm/L. . Hépatite B ou C. . Tuberculose active. Evaluation en ITT . Critère principal : ACR 20 à 24 semaines. . Critères secondaires : ACR 50 et ACR 70 à 24 semaines, critère HAQ, DAS 28.
Résultats
Nombre de malades 498 malades randomisés : Bras 1 : 175 - Bras 2 : 163 - Bras 3 : 160 Analyse des malades Pourcentage de malades ayant complété l’étude (24 semaines) : Bras 1 (tocilizumab 8 mg /kg) : 87 % (152) Bras 2 (tocilizumab 4 mg/kg) : 85 % (138) Bras 3 (placebo) : 79 % (127) Causes des sorties Les raisons de sortie d’étude les plus fréquentes étaient : les effets indésirables (11 malades du groupe tocilizumab 8 mg/kg, 10 malades du groupe tocilizumab 4 mg /kg, 10 malades du groupe contrôle) et l’insuffisance de réponse (4,6 et 19 malades respectivement). Profil des malades Les 3 groupes sont comparables Age moyen : 52 ans Pourcentage de femmes : 81 % Ancienneté de la PR (années) : 11,6 ans Nombre de DMARDs antérieurs : 2 CRP (mg/l) : 32 DAS 28 : 6,7 HAQ-DI : 1,7 …/…

-18-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau V (suite et fin). Etude RADIATE – étude de phase III randomisée, évaluant l’efficacité et la tolérance du tocilizumab dans la polyarthrite rhumatoïde présentant une réponse inadéquate aux anti-TNF-α (6) Résultats cliniques (suite) Efficacité
A S 24 Tocilizumab 8 mg/kg + MTX
Tocilizumab 4 mg/kg + MTX
MTX + placebo du tocilizumab
p
ACR 20 50 %* 30,4 % ** 10,1 % * et ** < 0,0001
ACR 50 28,8 % * 16,8 % ** 3,8 % * et ** <0,0001 ACR 70 12,4 % µ 5 % δ 1,3 % µ = 0,0001
δ = 0,1
DAS 28 < 3,2 à S 24 :
Bras 1 vs contrôle : 51,2 % vs 4,9 % Bras 2 vs contrôle : 15,2 % vs 4,9 % DAS 28 < 2,6 (rémission) à S 24 : Bras 1 vs contrôle : 30,1 % vs 1,6 % (p = 0,0001) bras 2 vs contrôle : 7,6 % vs 1,6 % (p= 0,053) Critère HAQ (variation par rapport à la valeur de base) Bras 1 vs contrôle : -0,39 vs -0,05 (p < 0,0001) Bras 2 vs contrôle : -0,31 vs -0,05 (p = 0,003) Amélioration statistiquement significative du handicap fonctionnel à S 24. Effets indésirables . La fréquence des effets indésirables est comparable dans les 2 groupes (84 % et 87,1% vs 80,6%) ; . Effets indésirables sévères : 13,7 % (bras 1), 13,5 % (bras 2), 19,4 % (bras 3). . Infections sévères : 4,6 % (bras 1) et 1,8 % (bras 2) vs 3,1% . Effets indésirables entraînant une interruption de traitement : 5,7 % (bras 1), 6,1 % (bras 2), 5 % (bras 3). . Effets indésirables les plus fréquemment observés : infections, troubles gastro-intestinaux, neutropénies, élévation du cholestérol. . ALAT > 3 LNS et < 5 LNS : 2 % (bras 1) et 2,5 % (bras 2) vs < 1% . HDL ≥ 60 mg/dL : 16,6 % (1) et 13,5 % (2) vs 3,8 % LDL ≥160 mg/dL : 12 % (1) et 15,3 % (2) vs 3,8% . Observation de neutropénies, de bas grade, non associées à fièvre ou infections, et transitoires.
Conclusion des auteurs
Les malades atteints de PR avec une réponse inadéquate ou une intolérance aux anti-TNF, répondent de façon effective à un traitement associant tocilizumab et méthotrexate avec un profil d’effets indésirables acceptable. Cette réponse est indépendante du nombre d’anti-TNF reçus antérieurement par le malade et est obtenue rapidement, dès la 2ème semaine.
Conclusion du CNHIM
Le tocilizumab a montré une efficacité en association au méthotrexate sur les signes et symptômes de PR active chez les malades présentant une réponse inadéquate ou une intolérance aux anti-TNFα. Cette étude porte sur de larges effectifs.

-19-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau VI. Etude OPTION – étude de phase III, randomisée, évaluant l’efficacité et la tolérance du tocilizumab dans la polyarthrite rhumatoïde présentant une réponse inadéquate au méthotrexate (7)
Etude OPTION. TOcilizumab Pivotal Trial in methotrexate Inadequate responders – 2008 (7).
Méthodologie
Objectif Evaluer l’efficacité et la tolérance du tocilizumab chez des malades atteints de PR active modérée à sévère, présentant une réponse inadéquate au méthotrexate. Type d’étude Etude randomisée, de phase III, contrôlée, en double aveugle contre placebo, internationale (73 centres dans 17 pays). Schéma et posologie 3 bras de traitement : Bras 1 : tocilizumab 8 mg/kg par voie IV, toutes les 4 semaines, associé au méthotrexate. Bras 2 : tocilizumab 4 mg/kg par voie IV, toutes les 4 semaines, associé au méthotrexate. Bras 3 (contrôle) : placebo par voie IV, toutes les 4 semaines, associé au méthotrexate. Méthotrexate : 10 à 25 mg par semaine. A 16 semaines, en l’absence d’au moins 20 % d’amélioration sur le nombre d’articulations gonflées et douloureuses, mise en place d’un traitement de secours : . pour le bras 1 : injection intra-articulaire de corticoïdes ou augmentation des corticoïdes oraux (maximum 10 mg/jour) ; . pour les bras 2 et 3, passage au tocilizumab 8 mg/kg par voie IV toutes les 4 semaines. Durée : 24 semaines.
Inclusion – Evaluation
Inclusion - Malades adultes atteints de PR, dont le diagnostic date d’au moins 6 mois et dont la réponse clinique au méthotrexate est inadéquate. - PR selon critères ACR, active : . nombre d’articulations gonflées ≥ 6 et nombre d’articulations douloureuses ≥ 8, . CRP ≥ 10 mg/L, . VS > 28 mm/h. - Malades sous méthotrexate durant les 12 dernières semaines à une dose de 10 à 25 mg, stable depuis au moins 8 semaines. - Corticoïdes à des doses inférieures ou égales à 10 mg/jour (prednisone ou équivalent), stables depuis au moins 6 semaines. Exclusion . Autre maladie auto-immune ou complication systémique de PR (vascularite, fibrose pulmonaire). . PR au stade IV fonctionnel. . Autre pathologie inflammatoire ou antécédent d’autre pathologie inflammatoire. . Infections récurrentes. . Fonction hépatique anormale : ALAT et ASAT > 1,5 LNS. . Antériorité de traitement inefficace ou non toléré par anti-TNF. Evaluation Critère principal : ACR 20 à 24 semaines. Critères secondaires : . ACR 50 et ACR 70 à 24 semaines ; . DAS 28 ; . évaluation de la fonction physique (HAQ-DI) ; . variation du taux moyen d’Hb, critères individuels ACR (variation du nombre d’articulations gonflées et douloureuses, CRP, VS…), fatigue (score FACIT) et qualité de vie (SF 36).
Résultats
Nombre de malades 623 malades randomisés : bras 1 = 205 ; bras 2 = 214 ; bras 3 = 204.
Analyse des malades : 622 malades (1 malade du bras 2 a reçu du tocilizumab 8 mg/kg). Bras 1 : 205, dont 14 sorties avant la fin de l’étude, et 19 malades ayant bénéficié d’une Rescue Therapy. Bras 2 : 213, dont 27 sorties avant la fin de l’étude, et 31 malades ayant bénéficié d’une Rescue Therapy. Bras 3 : 204, dont 15 sorties avant la fin de l’étude, et 68 malades ayant bénéficié d’une Rescue Therapy.
Profil des malades : Les 3 groupes sont comparables. Selon les bras de traitement : . Age moyen : 50,6 ans . Poids moyen : de 68 à 71,6 kg . Ancienneté de la pathologie : de 7,4 à 7,8 ans . Nombre de DMARDs reçus avant la randomisation : de 1,5 à 1,7 . % de malades ayant été traité antérieurement par anti-TNF : de 5 à 10 % . DAS 28 : 6,8 . CRP : de 24 à 28 mg/L . HAQ DI score : de 1,5 à 1,6 . FACIT-fatigue score : de 26,7 à 27 Quel que soit le bras de traitement : prédominance fémi-nine (autour de 80 %).
Résultats cliniques Efficacité ACR 20 à 24 semaines : Bras 1 vs contrôle : 59 % vs 26 % p < 0,0001 Bras 2 vs contrôle : 48 % vs 26 % p < 0,0001 ACR 50 à 24 semaines : Bras 1 vs contrôle : 44 % vs 11 % p < 0,0001 Bras 2 vs contrôle : 31 % vs 11 % p < 0,0001 ACR 70 à 24 semaines : Bras 1 vs contrôle : 22 % vs 2 % p < 0,0001 Bras 2 vs contrôle : 12 % vs 2 % p < 0,0001 …/…

-20-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau VI (suite et fin). Etude OPTION – étude de phase III, randomisée, évaluant l’efficacité et la tolérance du tocilizumab dans la polyarthrite rhumatoïde présentant une réponse inadéquate au méthotrexate (7)
Résultats (suite) Réponse EULAR :
Bonne Modérée Absente
Bras 1 vs contrôle : 38 % vs 3 % p < 0,0001
Bras 1 vs contrôle : 41 % vs 32 % p < 0,0001
Bras 1 vs contrôle : 20 % vs 65 % p < 0,0001
Bras 2 vs contrôle : 21 % vs 3 % p < 0,0001
Bras 2 vs contrôle : 41 % vs 32 % p < 0,0001
Bras 1 vs contrôle : 38 % vs 65 % p < 0,0001
DAS 28 < 2,6 = Rémission Bras 1 vs contrôle : 27 % vs 0,8 % p < 0,0001 Bras 2 vs contrôle : 13 % vs 0,8 % p = 0,0002 Composants individuels de la réponse ACR : - La CRP s’est normalisée dès la 2ème semaine dans le bras tocilizumab 8 mg/kg (p<0,0001 vs valeur initiale) et non dans le bras tocilizumab 4 mg/kg. - Une amélioration significative du score HAQ et de la fatigue a été observée dans les deux bras tocilizumab par rapport au bras contrôle. - Une augmentation du taux d’hémoglobine de 0,6-0,7 g/dL a été observée au bout de 4 semaines dans les deux bras tocilizumab (le taux d’Hb étant significativement plus élevé à S 24 versus placebo).
Effets indésirables . Taux des infections : 101,9 pour 100 malades-année bras 1 : 98,7 dans le bras 2, 96,2 dans le bras placebo. . Effets indésirables les plus fréquents : effets cutanés ; variations biologiques des paramètres suivants : ASAT, ALAT, polynucléaires neutrophiles et cholestérol ; troubles gastro-intestinaux (les pourcentages de survenue de 22, 23 et 22 % respectivement dans les groupes tocilizumab 8 mg/kg, 4 mg/kg et contrôle). . Effets indésirables sévères : 13 cas dans le bras 1, 13 cas dans le bras 2, et 12 dans le bras 3.
Conclusion des auteurs
Cette étude dont l’objet n’était pas de comparer les 2 doses de tocilizumab, met en évidence l’efficacité des doses de 4 et de 8 mg/kg chez des malades atteints de PR active modérée à sévère ayant une réponse inadéquate au méthotrexate. Le nombre de sorties d’étude a été plus important dans le groupe tocilizumab 4 mg/kg. L’amélioration clinique survient 2 à 4 semaines après l’initiation du traitement. La CRP se normalise après 2 semaines de traitement. Près d’un quart des malades sous tocilizumab 8 mg/kg sont en rémission contre moins de 1 % dans le groupe placebo.
Conclusion du CNHIM
Cette étude a porté sur de larges effectifs. Contrairement à l’étude CHARISMA dont la durée était plus courte (16 semaines avec les médicaments expérimentaux, mais étude de phase IIb), l’étude OPTION a pu montrer sur une durée d’évaluation habituelle dans la PR (24 semaines) une efficacité du tocilizumab l’effet du tocilizumab à 4 et 8 mg/kg (4 mg/kg = AMM aux USA) plus méthotrexate, en association au méthotrexate sur les signes et symptômes de PR active chez les malades présentant une réponse inadéquate ou une intolérance au méthotrexate. Le taux de réponse ACR 20 est significativement supérieur au bras contrôle. La réponse est rapidement observée, dès la 2ème semaine. Le profil de tolérance a été évalué sur 623 malades. Les effets indésirables les plus fréquemment observés sont : infections, neutropénies, élévations des enzymes hépatiques ALAT et ASAT, élévation du cholestérol, troubles gastro-intestinaux, réactions cutanées et réactions à l’injection. Cette étude n’évalue pas l’action du tocilizumab sur la progression structurale.

-21-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau VII. Etude TOWARD – étude de phase III, randomisée, évaluant l’efficacité et la tolérance du tocilizumab par rapport au placebo, en association à un ou plusieurs traitements de fond conventionnels (8)
Etude TOWARD. Tocilizumab in cOmbination With traditional DMARD Therapy – 2008 (8).
Méthodologie
Objectif Evaluer l’efficacité et la tolérance du tocilizumab par rapport au placebo, en association à un ou plusieurs traitements de fond conventionnels (DMARDs). Type d’étude Etude de phase III, internationale, randomisée, contrôlée, en double-aveugle contre placebo. Schéma et posologie 2 bras de traitement : Bras 1 : placebo + DMARD ; Bras 2 : tocilizumab 8 mg/kg + DMARD. Tocilizumab / placebo : une injection par voie IV, toutes les 4 semaines. Durée : 24 semaines
Inclusion – Evaluation
Inclusion - Malades adultes avec une PR ≥ 6 mois diagnostiquée selon les critères ACR (nombre d’articula-tions gonflées ≥ 6 et nombre d’articulations douloureuses ≥ 8, CRP ≥ 10 mg/L, VS ≥ 28 mm/h). - Malades ayant des doses stables de DMARDs (méthotrexate, chloro-quine, hydroxychloroquine, sels d’or, sulfasalazine, azathioprine et léflunomide) dans les 8 dernières semaines. - Corticoïdes (à des doses inférieures à 10 mg/j) et les AINS sont autorisés si la dose est stable depuis au moins 6 semaines. Exclusion Malades en échec d’un traitement par un anti-TNF et malades ayant été traité par une thérapie anti-cellulaire. Evaluation Critère principal : ACR 20 à 24 semaines. Critères secondaires : ACR 50 et ACR 70 à 24 semaines, critère HAQ, FACIT-F, DAS 28 à 24 semaines, variation de l’hémoglobine.
Résultats
Nombre de malades 1220 malades randomisés : Bras 1 : 415, Bras 2 : 805 Analyse des malades Pourcentage de malades ayant complété l’étude (24 semaines) : Bras 1 : 89 % (370) Bras 2 : 93 % (751) Causes des sorties : NR Profil des malades Les 2 groupes sont comparables : Age moyen: 53,5 ans. Poids : 73 ,5 kg. Pourcentage de femmes : 82,5 % Ancienneté de la maladie (années) : 9,8. CRP (mg/L) : 26. DAS 28 : 6,7. HAQ-DI: 1,5. Dose (mg) de MTX /semaine : 15.
…/…

-22-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau VII (suite et fin). Etude TOWARD – étude de phase III, randomisée, évaluant l’efficacité et la tolérance du tocilizumab par rapport au placebo, en association à un ou plusieurs traitements de fond conventionnels (8)
Résultats cliniques (suite) Efficacité
S 24 Placebo + DMARD (bras 1)
tocilizumab + DMARD (bras 2)
p
ACR 20 25 % 61 % < 0,0001 ACR 50 9 % 38 % < 0,0001 ACR 70 3 % 21 % < 0,0001
DAS 28 < 3,2 à S 24 : 45 % (bras 2) vs 6 % (bras 1) p < 0,0001 DAS 28 < 2,6 à S 24 (rémission) : 30 % (bras 2) vs 3 % (bras 1) p < 0,0001 Réponse EULAR à S 24 : 80 % (bras 2) vs 38 % (bras 1) p < 0,0001 Critère HAQ (variation par rapport à la valeur de base) : Diminution de –0,5 (bras 2) vs –0,2 (bras 1) p < 0,001 Critère FACIT-F (variation par rapport à la valeur de base) : Amélioration de 8 (bras 2) vs 3,6 (bras 1) p < 0,001 Score SF–36 (variation par rapport à la valeur de base) : sur le plan physique, amélioration 8,9 % (bras 2) vs 4,1 % (bras 1) p < 0,001 sur le plan mental, amélioration 5,3 % (bras 2) vs 2,3 % (bras 1) p < 0,001 Effets indésirables (EI) - EI : 72,8 % (bras 2) vs 61,1% (bras 1) - EI sévères : 6,7 % (bras 2) vs 4,3 % (bras 1) - EI conduisant à un arrêt du traitement : 3,9 % (bras 2) vs 1,9 % (bras 1) - Infections sévères : 2,7 % (bras 2) vs 1,9 % (bras 1) - L’augmentation des transaminases a été plus fréquente dans le groupe tocilizumab ; dans la majorité des cas, cette élévation (maximale 2 semaines après la perfusion) restait inférieure à 3 fois les LSN ; 3 arrêts ont été observés dans le groupe tocilizumab (bras 2), l’un en raison d’une élévation des ALAT, les deux autres pour élévation de la bilirubine). - Augmentation du cholestérol total à S 24 (cholestérol total ≤ 2,4 g/L à l’initiation et ≥ 2,4 g/L à S 24) 23 % des malades (bras 2) vs 5,5 % (bras 1) - LDL–cholestérol à S 24 (LDL ≤ 1,6 g/L à l’initiation et ≥ 1,6 g/L à S 24): 16 % des malades (bras 2) vs 3 % (bras 1) - HDL-cholestérol (HDL ≤ 0,6 g/L à l’initiation et ≥ 0,6 g/L à S 24): 15 % des malades (bras 2) vs 6 % (bras 1) - Triglycérides (TG ≤ 0,5 g/L à l’initiation et ≥ 0,5 g/L à S 24) 1,4 % des malades (bras 2) vs 1 % (bras 1) - Polynucléaires neutrophiles (PN) : Une proportion plus importante de malades sous tocilizumab présente une baisse des PN par rapport au groupe placebo (majoritairement des neutropénies de grade 1 et 2).
Conclusion des auteurs
Le tocilizumab en association à un traitement par DMARD classique a montré une diminution significative et rapide de l’activité de la PR par rapport au placebo. Sur le plan de la tolérance, les augmentations du taux de cholestérol et des enzymes hépatiques ne sont pas associées à des symptômes cliniques. Aucune corrélation ne semble exister entre la baisse du taux de PN et la survenue d’infections.
Conclusion du CNHIM
En association aux traitements de fond classiques, le tocilizumab a un effet supérieur au placebo. Sur le plan de la tolérance, des augmentations du cholestérol et des enzymes hépatiques sont observées dans le bras tocilizumab, il conviendra de les dépister et de les prendre en charge.

-23-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau VIII. Etude AMBITION – étude randomisée versus placebo, évaluant l’efficacité et la tolérance du tocilizumab en monothérapie par rapport au méthotrexate en monothérapie dans la polyarthrite rhumatoïde active sans échec antérieur au méthotrexate ou à une biothérapie (9)
Etude AMBITION. ACTEMRA versus Methotrexate double-Blind Investigative Trial In mONotherapy – 2010 (9).
Méthodologie
Objectif Evaluer l’efficacité et la tolérance du tocilizumab en monothérapie par rapport au méthotrexate en monothérapie chez des malades atteints de PR active n’ayant pas présenté antérieurement d’échec au méthotrexate ou à une biothérapie.
Type d’étude Etude randomisée, contrôlée, en double-aveugle contre placebo.
Schéma et posologie 2 bras de traitement : Bras 1 : Méthotrexate per os Escalade de dose selon le schéma suivant : 7,5 mg/semaine puis 15 mg/sem à partir de la semaine 4, puis 20 mg/semaine à partir de la semaine 8. L’administration de méthotrexate est associée à la prise de folates à une dose ≥ 5 mg/sem. Pour des raisons de tolérance, il est permis de diminuer la dose hebdomadaire de méthotrexate à 10 mg. Associé au placebo du tocilizumab en 1 perfusion toutes les 4 semaines.
Bras 2 : tocilizumab sous forme d’une perfusion de 8 mg/kg toutes les 4 semaines, associé au placebo du méthotrexate 1 fois par semaine. Dans le but de tester la sensibilité pour la non-infériorité, une sous-étude a été menée aux USA, Israël et Canada. Elle comprend 3 bras (1 :1 :1) : les 2 bras cités ci-dessus et un bras supplémentaire. Dans ce dernier, les malades reçoivent : le placebo du méthotrexate et le placebo du tocilizumab pendant les 8 premières semaines (soit 2 perfusions) ; à partir de la semaine 8, ils reçoivent une perfusion de tocilizumab 8 mg/kg toutes les 4 semaines pendant les 16 semaines restantes de l’étude, à la place du placebo. Les malades sont susceptibles de recevoir un traitement de secours de tocilizumab 8 mg/kg avant la semaine 8 si l’investigateur le juge nécessaire au vu du nombre d’articulations gonflées et douloureuses.
Durée : 24 semaines
Inclusion – Evaluation
Inclusion - Malades adultes atteints de PR, modérée à sévère depuis au moins 3 mois. - PR selon critères ACR, active : . articulations gonflées ≥ 6 et douloureuses ≥ 8 ; . CRP ≥ 10 mg/L ; . VS > 28 mm/h. - Corticoïdes à des doses inférieures ou égales à 10 mg/jour (prednisone ou équivalent), stables depuis 6 mois. Exclusion . Pathologie instable concomi-tante. . Tuberculose latente, active ou non traitée. . Malades antérieurement traités sans succès par anti-TNFα. . Malades ayant reçu du méthotrexate dans les 6 mois précédant la randomisation. . Malades traités antérieurement par méthotrexate et l’ayant arrêté pour effets indésirables ou pour inefficacité. Evaluation Critère principal : ACR 20 à 24 semaines. Critères secondaires : ACR 50 et ACR 70 à 24 semaines, DAS 28, réponse Eular, HAQ-DI, évolution du taux d’hémo-globine. Etude de non-infériorité contre méthotrexate en Per Protocole (PP), avec étude de supériorité en ITT en cas d’atteinte de la non-infériorité.
Résultats Nombre de malades 673 malades : Bras 1 (méthotrexate) 284 malades – Bras 2 (tocilizumab) 288 malades – Bras supplémentaire sous-étude (placebo puis tocilizumab) : 101 Analyse des malades Pourcentage des malades ayant complété l’étude (S 24) : Bras 1 : 262 malades (92 %) Bras 2 : 268 (93 %) Bras supplémentaire : 82 (81%) Profil des malades Les 2 groupes sont comparables Age moyen: 50,3 ans Poids : NR Pourcentage de femmes : 80% Ancienneté de la maladie : 6,3 ans CRP (mg/L) : 30 DAS 28 : 6,8 HAQ-DI: 1,6 Pourcentage de malades naïfs de méthotrexate : 67 % par bras Pourcentage de malades ayant reçu des anti-TNFα: environ 8 % Résultats cliniques Efficacité clinique . Mise en évidence de la non-infériorité sur la population PP pour le critère principal (bras tocilizumab n = 265 ; bras méthotrexate n = 259) Le critère ACR 20 à S 24 est atteint par 70,6 % des malades sous tocilizumab contre 52,1 % sous méthotrexate. …/…

-24-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau VIII (suite et fin). Etude AMBITION - étude randomisée versus placebo, évaluant l’efficacité et la tolérance du tocilizumab en monothérapie par rapport au méthotrexate en monothérapie dans la polyarthrite rhumatoïde active sans échec antérieur au méthotrexate ou à une biothérapie (9)
Résultats cliniques (suite)
Résultats en ITT : supériorité démontrée pour le groupe tocilizumab
S 24 Bras 1 (méthotrexate) Bras 2 (tocilizumab) p
ACR 20 52,5 % 69,9 % p < 0,001 ACR 50 33,5 % 44,1 % p = 0,002 ACR 70 15,1 % 28 % p < 0,001
A S 24, les malades du groupe tocilizumab ont 5 fois plus de chance d’être en rémission (DAS 28 < 2,6) : « odds ratio » versus méthotrexate = 5,83. Ces mêmes malades ont 4 fois plus de chance d’atteindre une réponse Eular au moins modérée : « odds ratio » versus méthotrexate = 4,24. . Analyse en sous-groupes : le tocilizumab 8 mg /kg est supérieur au méthotrexate aussi bien chez les malades naïfs de méthotrexate que chez les malades en ayant déjà reçu, pour les réponses ACR 20, ACR 50, et ACR 70 à 24 semaines. . A S 24, les taux d’hémoglobine augmentent de 1,19 g/dL par rapport à la valeur initiale dans le groupe tocilizumab, de 0,10 g/dL dans le bras méthotrexate. Dans le groupe tocilizumab, l’amélioration est observée précocement, dès la 2ème semaine. A S6, le taux d’hémoglobine se normalise chez les malades sous la limite de la normale à l’inclusion, et se maintient jusqu’à S 24. . Score HAQ-DI (évolution par rapport à la valeur initiale) Groupe tocilizumab : - 0,7 Groupe méthotrexate : - 0,5 Amélioration de la fonction physique en faveur du tocilizumab.
Effets indésirables Le taux global d’effets indésirables (EI) a été similaire dans les deux groupes, de même que le taux d’effets indésirables graves (79,9 % dans le groupe tocilizumab contre 77,5 ù dans le groupe méthotrexate, p – 0,484). Le type d’EI le plus fréquent est l’infection : 34,4 % dans le groupe tocilizumab contre 37,3 % dans le groupe méthotrexate. Des infections sévères ont été rapportées pour 4 malades dans le groupe tocilizumab et 2 dans le groupe méthotrexate (p=0,422). En termes de fréquence, les désordres gastro-intestinaux sont en seconde place, avec une fréquence similaire dans les 2 groupes. Les réactions liées à la perfusion surviennent chez 5,6 % des malades sous tocilizumab. La majorité de ces réactions surviennent pendant les deux premières perfusions. Quatre décès ont été observés, un dans le groupe méthotrexate (cancer du poumon), trois dans le groupe tocilizumab (ischémie myocardique, arrêt cardiorespiratoire et perforation/hémorragie gastro-intestinale haute). Les neutropénies sont plus fréquentes dans le groupe tocilizumab, 3,1 % contre 0,4 % dans le groupe méthotrexate. Elles sont de grade 3 et réversibles.
Conclusion des auteurs
L’étude AMBITION démontre l’efficacité du tocilizumab en monothérapie par rapport à un traitement de méthotrexate, chez des malades dont le diagnostic de la maladie est relativement peu ancien, sans échec antérieur au méthotrexate. A court terme, deux effets indésirables semblent différencier le tocilizumab du méthotrexate : les élévations du cholestérol total, du cholestérol-HDL et du cholestérol-LDL, et les neutropénies réversibles.
Conclusion du CNHIM
La supériorité du tocilizumab 8 mg/kg en monothérapie a été démontrée par rapport au méthotrexate en monothérapie utilisé comme comparateur, quelle que soit l’exposition antérieure des malades au méthotrexate.

-25-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
L’étude LITHE (TociLIzumab safety and THE prevention of structural joint damage) (10) avait pour objectifs, chez des malades atteints d’une PR modérée à sévère depuis plus de 6 mois ayant une réponse inadéquate au méthotrexate, d’évaluer : - l’efficacité et la tolérance du tocilizumab par rapport au placebo, en association au méthotrexate, à 24 semaines ;
- l’efficacité sur le ralentissement de la destruction articulaire sur une période de 52 semaines ;
- l’amélioration de la fonction physique sur une période de 52 semaines.
La réduction de la progression des dommages structuraux articulaires a été évaluée par radiographie et exprimée par le changement du score total de Sharp modifié par Genant et ses composants, le score d’érosion et le score de pincement articulaire.
Il s’agit d’une étude de phase III, randomisée, contrôlée, en double-aveugle, pendant 52 semaines, poursuivie en ouvert la deuxième année avec une phase d’extension optionnelle de 3 ans. Trois 3 bras de traitement ont été retenus : Bras 1 : tocilizumab 8 mg/kg par voie IV toutes les 4 semaines Bras 2 : tocilizumab 4 mg/kg par voie IV toutes les 4 semaines Bras placebo par voie IV toutes les 4 semaines. Le traitement associé comprenait le méthotrexate, à la posologie de 10 à 25 mg/semaine, débuté au moins 12 semaines avant la randomisation. A partir de la semaine 16, les malades insuffisamment répondeurs (moins de 20 % d’amélioration des NAD/NAG) pouvaient recevoir un traitement de secours par tocilizumab. A la fin de la première année, les malades avec moins de 70 % d’amélioration des NAD/NAG, continuaient l’étude sous tocilizumab 8 mg /kg en ouvert (les autres pouvaient : - soit poursuivre l’étude en double aveugle, - soit passer en ouvert).
Les critères principaux pour l’évaluation ont été : - la variation du score total de Sharp modifié par Genant (extrapolation linéaire pour les données manquantes, les données post-
traitement de secours étant considérées comme man-quantes) et – la variation du score HAQ (en AUC) à 2 ans.
Les résultats obtenus ont été les suivants :
. le profil des malades : PR, d’ancienneté moyenne égale à 9 ans, un DAS28 moyen à l’inclusion de 6,5 ;
. la réduction de la progression des dommages structuraux a été mise en évidence par un taux de progression radiographique significativement moindre chez les malades traités par tocilizumab comparativement au groupe contrôle.
A 52 semaines, l’effet du tocilizumab à 8 mg/kg (effet déjà démontré à 4 mg/kg = AMM aux USA) plus méthotrexate, sur le ralentissement de la destruction articulaire, a été statistiquement démontré par rapport au groupe placebo : Score total de Sharp modifié par Genant de 0,29 (groupe tocilizumab) versus 1,13 (p < 0,001).
A 2 ans, les malades traités sous tocilizumab 8 mg/kg ont eu significa-tivement moins de progression radiologique (81 % d’inhibition) par rapport aux malades sous méthotrexate (10). - Score total de Sharp : Placebo + MTX (n = 393) = + 1,96 Tocilizumab 8 mg/kg + MTX (n = 398) = + 0,37 - Pourcentage de malades ne progressant pas : Placebo + MTX (n = 393) = 66 % Tocilizumab 8 mg/kg + MTX (n = 398) = 83 %* - HAQ (AUC) : Placebo + MTX (n = 393) = -139,4 Tocilizumab 8 mg/kg + MTX (n = 398) = -320,8* * p <0,0001
Efficacité clinique à 1 an : ACR20 - 24 semaines : Bras 1 : 56,3 % Bras 2 : 50,6 % Placebo : 27 % p < 0,0001
- 52 semaines : Bras 1 : 56 % Bras 2 : 47 % Placebo : 25 % p < 0,0001
A 2 ans, le taux de malades avec une réponse ACR 70 était plus élevé sous tocilizumab 8 mg/kg plus métho-trexate que sous méthotrexate plus placebo.
Le taux de malades en rémission (DAS 28 inférieur à 2,6) sous tocilizumab 8 mg/kg + méthotrexate était de 65 % (156 malades sur 241) à S 104 contre 48 % (132/275) à la fin de la première année.
Dans toutes les études développées ci-dessus, le délai d’apparition de la réponse au tocilizumab a été rapide (dès la 2ème semaine) et l’amplitude de la réponse a continué à augmenter au cours du traitement. Des réponses durables et continues ont été observées pendant plus de 24 mois au cours des études d’extension en ouvert AMBITION, OPTION, TOWARD et RADIATE.
La proportion de malades ayant atteint une rémission à 24 semaines (DAS 28 < 2,6) a été significa-tivement supérieure chez les malades recevant le tocilizumab (28 à 34 %) par rapport aux malades du groupe contrôle (1 à 12 %).
Les malades traités par tocilizumab ont une amélioration de leurs capacités fonctionnelles dans les questionnaires d’auto-évaluation (questionnaire d’évaluation de l’état de santé : HAQ-DI, SF-36 et fatigue mesurée à l’aide des scores d’évaluation fonctionnelle du traitement de maladies chroniques : FACIT-Fatigue). Des améliorations statistiquement significatives du HAQ-DI ont été observées chez les malades traités par ROACTEMRA® par rapport aux malades ayant reçu des DMARDs.
L’étude STREAM (11), étude d’extension en ouvert sur 5 ans, a prolongé une étude initiale en aveugle d’une durée de 24 semaines. Les malades ont reçu une perfusion mensuelle de tocilizumab à la dose de 8 mg/kg en monothérapie.
Cent quarante trois malades ont été inclus et 94 d’entre eux ont complété les 5 ans de suivi (soit 66 %). A 5 ans, 84 % des malades ont une réponse à l’ACR 20, 69,1 % à l’ACR 50 et 43,6 % à l’ACR 70. 55,3 % des malades ont atteint la rémission avec un score de DAS 28 inférieur à 2,6.
Sur 612 malades-année, le taux d’effets indésirables a été estimé à 27,5 évènements pour 100 malades-année, le taux d’infections sévères à 5,7 évènements pour 100 malades-année. Quatre cas de tumeurs ont été observés. Ces résultats sont en faveur d’une efficacité qui se

-26-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
maintient dans le temps avec un profil de tolérance acceptable.
2.4. Renseignements théra-peutiques
Cf. tableau IX.
2.4.1. Posologie et mode d’administration (42)
La spécialité ROACTEMRA® est une
solution à diluer pour perfusion contenant 20 mg/mL de tocilizumab. Elle se présente à 3 dosages différents : 4 mL (80 mg), 10 mL (200 mg) et 20 mL (400 mg). Après dilution, le médicament doit être administré par perfusion intravei-neuse d’une durée d’une heure.
La posologie recommandée est de 8 mg/kg soit 0,4 mL/kg. Cette posologie peut être adaptée dans certaines situations (anomalies des paramètres biologiques) : 4 mg/kg. La dose maximale recommandée est de 800 mg par perfusion. Des doses supérieures à 800 mg ne sont pas recommandées chez des sujets ayant un poids supérieur à 100 kg (42).
La dilution est réalisée à l’aide d’une solution de chlorure de sodium à 0,9 % pour atteindre un volume final de 100 ml. Le débit d’administration doit être contrôlé comme suit : - 10 mL/h pendant 15 minutes, - puis 130 ml/h pendant 45 minutes.
2.4.2. Effets indésirables (2, 42, 43)
Cf. tableau X. Au cours du développement clinique, 3 778 malades ont reçu au moins une dose de tocilizumab à une posologie de 4 mg/kg ou de 8 mg/kg.
Les effets indésirables présentés dans le tableau sont issus de l’analyse de la tolérance du tocilizumab :
- en association au méthotrexate (ou à un autre DMARD) au cours de 4 études contrôlées contre placebo associé au méthotrexate (ou à un DMARD) : études LITHE (10), OPTION (7), TOWARDS (8), et RADIATE (6) ;
- en monothérapie au cours d’une étude contrôlée versus métho-trexate : étude AMBITION (9).
La répartition des 2 644 malades, au cours de ces études, a été la suivante : - 774 malades ont reçu le tocilizumab à la posologie de 4 mg/kg en association au méthotrexate ; - 1 582 malades ont reçu le tocilizumab à la posologie de 8 mg/kg en association au métho-trexate ou à un autre DMARD ; - 288 malades ont reçu le tocilizumab à la posologie de 8 mg/kg en monothérapie.
Les études d’extension en ouvert à long terme ont inclus 2 562 malades ayant reçu le tocilizumab à la posologie de 8 mg/kg avec ou sans DMARD associé. L’exposition totale dans l’analyse de la tolérance à long terme a été de 3 685 malades-années.
Les effets indésirables les plus fréquemment rapportés (c'est-à-dire, survenus chez plus de 5 % des malades traités par tocilizumab en monothérapie ou en association à un DMARD) ont été les suivants :
- infections des voies respiratoires supérieures ; - rhinopharyngite ; - céphalées ; - hypertension ; - augmentation des ALAT.
Cas particulier des infections : Au cours des études contrôlées, le taux global des infections rapportées avec le tocilizumab 8 mg/kg associé à un DMARD a été de 127 évènements pour 100 malades-années, par rapport à 112 évènements pour 100 malades-années dans le groupe placebo associé à un DMARD.
Dans la population étudiée à long terme, le taux d’infection avec le tocilizumab associé à un DMARD a été de 108 évènements pour 100 malades-années.
Au cours des études cliniques contrôlées, le taux d’infections graves sous tocilizumab 8 mg/kg associé à un DMARD a été de 5,3 événements pour 100 malades-années par rapport à 3,9 événements pour 100 malades-années dans le groupe placebo associé à un DMARD.
Dans l’étude en monothérapie, le taux d’infections graves observées avec le tocilizumab associé à un DMARD à été de 3,9 événements pour 100 malades-années.
Les infections graves rapportées ont été les suivantes : pneumonie, cellulite, zona, gastro-entérite, diverticulite, septicémie et arthrite bactérienne.
Les infections graves ont rarement été fatales. Des cas d’infections opportunistes ont été rapportés.
Complications de diverticulite : Pendant les six mois des études contrôlées, des complications de diverticulites, de type péritonite purulente généralisée, perforation gastro-intestinale basse, fistule ou abcès, ont été rapportées sous tocilizumab mais peu fréquemment (0,1 % des malades).

-27-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau IX. Renseignements thérapeutiques du tocilizumab, ROACTEMRA® (42, 45)
Indication AMM Posologie Contre-indication
Traitement de la PR active, modérée à sévère, chez les malades adultes : En association au MTX, en cas de : - réponse inadéquate ou intolérant à au moins un traitement de fond (DMARDs) ; - réponse inadéquate ou intolérance à au moins un anti-TNF-α En monothérapie, en cas d’intolérance au MTX, lorsque la poursuite du traitement par MTX est inadaptée chez ces malades.
Il a été montré que ROACTEMRA®, en association avec le méthotrexate, réduit le taux de progression des dommages structuraux articulaires mesurés par radiographie et améliore les capacités fonctionnelles.
Une perfusion intraveineuse d’une heure de 8 mg/kg toutes les 4 semaines.
. Hypersensibilité à la substance active ou à l’un des excipients. . Infections sévères ou actives.
Tableau X. Effets indésirables du tocilizumab (6-10, 42)
Fréquence Pathologie
Très fréquent Fréquent Peu fréquent
Infections et infestations infection des voies respiratoires supérieures
cellulite, pneumonie, herpès labial, zona
diverticulite
Affections gastro-intestinales douleur abdominale, ulcère buccal, gastrite
Stomatite, ulcère gastrique
Affections de la peau et des tissus sous-cutanés
rash, prurit, urticaire
Affections du système nerveux céphalées, sensations vertigineuses
Investigations augmentation des transaminases hépatiques, prise de poids
augmentation de la bilirubine totale
Affections vasculaires hypertension
Affections hématologiques et du système lymphatique
leucopénie, neutropénie
Troubles du métabolisme et de la nutrition
hypercholestérolémie hypertriglycéridémie
Troubles généraux et anomalies au site d’administration
réactions d’hypersensibilité, œdème périphérique
Troubles respiratoires, thoraciques et médiastinaux
toux, dyspnée
Affections oculaires conjonctivite
Troubles rénaux néphrolithiase
Troubles endocriniens hypothyroïdisme

-28-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Réactions liées à la perfusion : Des événements indésirables associés à la perfusion (événements pendant ou dans les 24 heures suivant la perfusion) ont été rapportés chez 6,9 % des malades du groupe tocilizumab 8 mg/kg associé à un DMARD et 5,1 % des malades dans le groupe placebo associé à un DMARD. Les événe-ments décrits au cours des 24 premières heures suivant la perfusion ont été principalement : des épisodes d’hypertension, des céphalées et des réactions cutanées (rash, urticaire). Ils n’ont pas entraîné de modification du traitement.
Le taux des réactions anaphy-lactiques (survenues chez un total de 6 malades sur 3 778 soit 0,2 %) a été plus élevé avec la posologie de 4 mg/kg qu’avec celle de 8 mg/kg. Des réactions d’hyper-sensibilité clinique-ment significatives associées au tocilizumab et nécessitant l’interrup-tion du traitement ont été rapportées chez un total de 13 malades sur 3 778 (soit 0,3 %) traités par tocilizumab au cours des études cliniques contrôlées et en ouvert. Ces réactions ont été généralement observées entre la deuxième et la cinquième perfusion de tocilizumab.
Immunogénicité : La présence d’anticorps anti-tocilizumab a été recherchée chez un total de 2 876 malades au cours des études cliniques contrôlées. Quarante six malades (1,6 %) ont développé des anticorps anti-tocilizumab, parmi lesquels 6 ont présenté une réaction d’hypersensibilité médicalement significative ayant entraîné l’arrêt définitif du traitement chez 5 d’entre eux. Chez 30 malades (1,1 %) ayant développé des anticorps neutra-lisants, aucune corrélation apparente avec la réponse clinique n’a été observée.
Anomalies hématologiques : Une diminution du nombre de polynucléaires neutrophiles (PN) inférieure à 1 g/L a été observée chez 3,4 % des malades traités par tocilizumab 8 mg/kg en association à un DMARD par rapport à moins de 0,1 % des malades ayant reçu le placebo plus DMARD.
Approximativement la moitié des malades ayant présenté un nombre de neutrophiles inférieur à 1 G/L a développé cet effet indésirable dans les 8 semaines qui ont suivi le début
du traitement. Des diminutions inférieures à 0,5 G/L ont été rapportées chez 0,3% des malades recevant tocilizumab 8 mg/kg associé à un DMARD.
Aucune relation n’a été établie entre la diminution du nombre des neutrophiles et la survenue d’infections graves.
Une diminution de la numération plaquettaire inférieure à 100 000 /µL a été mise en évidence chez 1,7 % des malades recevant le tocilizumab 8mg/kg associé à un DMARD par rapport à moins de 1 % des malades sous placebo plus DMARD. Ces diminutions sont survenues sans événements hémorragiques associés.
Augmentation des transaminases hépatiques : Des augmentations des ALAT et ASAT supérieures à 3 fois les valeurs normales ont été observées chez 2,1 % des malades traités par tocilizumab 8 mg/kg par rapport à 4,9 % sous méthotrexate et chez 6,5 % des malades ayant reçu tocilizumab associé à un DMARD, par rapport à 1,5 % des malades sous placebo plus DMARD.
L’adjonction de médicaments poten-tiellement hépatotoxiques (par exemple, le méthotrexate) au tocilizumab a majoré la fréquence de ces augmentations.
Des élévations des ALAT et ASAT supérieures à 3 fois les valeurs normales ont été observées chez 0,7 % des malades recevant tocilizumab en monothérapie et chez 1,4 % des malades traités par tocilizumab plus DMARD. La majorité de ces malades ont interrompu définitivement le traitement par tocilizumab. Ces augmentations n’ont pas été associées à une élévation clini-quement significative de la bilirubine conjuguée, ni à des signes cliniques d’hépatite ou d’insuffisance hépati-que.
Paramètres lipidiques : Des élévations des paramètres lipidiques, tels que cholestérol total, triglycérides, LDL-cholestérol et/ou HDL-cholestérol, ont été rapportées fréquemment. Environ 24 % des malades recevant tocilizumab dans les études cliniques ont présenté des élévations prolongées du cholestérol total supérieures à 6,2 mmol/L, et 15 % ont présenté une élévation
prolongée du LDL-cholestérol supé-rieure à 4,1 mmol/L. Les augmentations des paramètres lipidiques ont répondu à un traite-ment par hypolipidémiant.
Affections malignes : Les données cliniques sont insuf-fisantes pour évaluer l’incidence potentielle des affections malignes après une exposition au tocilizumab.
2.4.3. Mises en garde et précautions d’emploi (42)
Le tocilizumab peut augmenter le risque d’infections et réactiver des infections latentes. Il ne doit pas être donné à des malades présentant une infection évolutive sévère et notamment une tuberculose active. Des précautions s’imposent chez les malades présentant une infection évolutive chronique ou ayant des antécédents d’infections récurrentes. Avant toute instauration de traitement, il convient de détecter toute infection grave, infection chronique et/ou récidivante ainsi que toute pathologie sous-jacente prédis-posant aux infections (diverticulite, diabète par exemple). Un dépistage de la tuberculose doit être réalisé. Toute tuberculose latente doit être traitée par l’association d’antituber-culeux avant de démarrer le tocilizumab.
Le tocilizumab doit être utilisé avec précaution chez les malades présen-tant des antécédents d’ulcération intestinale ou de diverticulite.
Le bilan biologique initial comprend en particulier l’évaluation des enzymes hépatiques (ASAT, ALAT). Leur dosage sera contrôlé toutes les 4 à 8 semaines pendant les 6 premiers mois de traitement, puis par la suite toutes les 12 semaines. Chez les malades présentant des augmentations supérieures à 1,5 fois la limite supérieure de la normale (LSN), la posologie du tocilizumab devra être réduite à 4 mg/kg. Dans le cas d’augmentations supérieures à 3 à 5 fois la limite supérieure de la normale, le traitement devra être interrompu jusqu’à ce que les transaminases soient inférieures à 3 fois la LSN. Chez les malades présentant une augmentation des ALAT ou ASAT supérieures à 5 fois la LSN, le traitement n’est pas recommandé.
Le bilan biologique comprend aussi un hémogramme initial puis toutes

-29-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
les 4 à 8 semaines après le début du traitement.
L’observation de neutropénies au cours des études cliniques a conduit aux recommandations d’adaptations posologiques suivantes : - si le nombre des neutrophiles est compris entre 0,5 et 1 G/L, le tocilizumab sera interrompu puis réintroduit lorsque les PN atteignent 1 G/L, à raison de 4 mg/kg dans un premier temps, puis à raison de 8 mg/kg si l’état clinique du malade le permet ; - si le nombre des PN est inférieur à 0,5 G/L, le traitement doit être arrêté.
La numération des plaquettes fait partie également du bilan initial et en contrôle toutes les 4 à 8 semaines après le début du traitement. Dans le cas d’une numération plaquettaire comprise entre 50 000 et 100 000 plaquettes par µl, le traitement doit être interrompu puis réintroduit après que le nombre de plaquettes soit remonté à une valeur égale ou supérieure à 100 000, à la posologie de 4 mg/kg puis de 8 mg/kg si l’état clinique du malade le permet. Toute numération plaquettaire en-dessous de 50 000 plaquettes contre-indique le traitement sous tocilizumab.
Un bilan lipidique initial et à la 8ème semaine puis en fonction des facteurs de risque associés seront réalisés. Il comprend le cholestérol total, le LDL-cholestérol, le HDL-cholestérol et les triglycérides. Des réactions graves d’hypersen-sibilité ont été rapportées lors de perfusions de tocilizumab. Un traitement approprié doit pouvoir être mis en œuvre immédiatement en cas de survenue d’une réaction anaphylactique au cours de l’administration de celui-ci.
2.4.4. Surdosage (42)
Les données disponibles sont limitées sur les conséquences d’un éventuel
surdosage par tocilizumab. Un cas accidentel a été rapporté, au cours duquel un malade atteint de myélome multiple a reçu une dose unique de 40 mg/kg. Aucune réaction indésirable n’a été observée.
De plus, aucune réaction indésirable grave n’a été observée chez des volontaires sains ayant reçu des doses uniques allant jusqu’à 28 mg/kg, en dehors d’un cas de neutropénie.
2.4.5. Interactions médica-menteuses (42)
L’expression des isoenzymes hépatiques du CYP 450 est supprimée par des cytokines, comme l’IL-6, qui stimulent l’inflammation chronique. Par conséquent, l’expression des isoenzymes du CYP 450 peut être restaurée lors de la mise en place d’un traitement entraînant une inhibition puissante des cytokines, comme le tocilizumab.
Lors de l’instauration ou lors de l’interruption d’un traitement par tocilizumab, les malades recevant des médicaments qui sont métabolisés par les isoenzymes CYP3A4, 1A2, ou 2C9 (par exemple, l’atorvastatine, les inhibiteurs calciques, la théophylline, la war-farine, la phénytoïne, la ciclosporine ou les benzodiazépines) nécessitant des ajustements individuels, doivent être contrôlés dans la mesure où la posologie peut devoir être augmentée afin de maintenir l’effet thérapeutique.
Compte tenu de sa demi-vie d’élimination relativement longue (t1/2), l’effet du tocilizumab sur l’activité des enzymes du CYP450 peut persister plusieurs semaines après l’arrêt du traitement.
Il ne faut pas administrer de vaccins vivants et de vaccins atténués simultanément à un traitement par tocilizumab.
2.4.6. Grossesse et allai-tement (42)
Il n’existe pas de données suffisamment pertinentes concernant l’utilisation du ROACTEMRA® chez la femme enceinte. Les femmes en âge de procréer doivent utiliser une contraception efficace pendant le traitement par tocilizumab et jusqu’à 3 mois suivant son arrêt. En ce qui concerne l’allaitement, il est recommandé par mesure de prudence, d’attendre au moins 3 mois après le dernier traitement par tocilizumab avant de le débuter.
2.5. Plan de gestion des risques et cohortes de suivi
En complément du circuit de pharmacovigilance classique, un plan de gestion de risque (PGR) a été mis en place au niveau européen. Il comprend des mesures d’évaluation des risques, en particulier : - une surveillance spécifique des effets indésirables d’intérêt particulier comme les infections graves ; - des registres pour mieux évaluer le profil de sécurité du RoActemra®, notamment à long terme.
Des outils de minimisation du risque sont mis à disposition des profes-sionnels de santé. Une carte de surveillance est remise au malade.
A côté du PGR européen, le registre REGATE (REGistre AcTEmra) a été mis en place en France sous l’égide de la Société Française de Rhumatologie (SFR) et du Club Rhumatismes et Inflammation (CRI). Il permet l’étude de l’utilisation du tocilizumab dans la vie réelle. Comme pour les biothérapies déjà commercialisées, le CRI a élaboré des fiches pratiques pour aider les praticiens dans leur démarche d’instauration et de suivi de traitement (bilan pré-thérapeutique, le suivi thérapeutique et les effets indésirables d’un traitement par tocilizumab).

-30-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
3. Certolizumab
3.1. Renseignements gé-néraux et galéniques (42) Cf. tableau XI.
3.2. Propriétés pharmaco-logiques
3.2.1. Mode d’obtention et mode d’action Le certolizumab pegol est un fragment Fab’ d’anticorps humanisé recombinant, dirigé contre le TNF-α, exprimé dans Escherichia coli, et conjugué à deux molécules de polyéthylène glycol (PEG). L’obtention du certolizumab se fait par la technique de la fermentation microbienne. La première étape du développement du certolizumab pegol consiste à sélectionner l’hybridome spécifique de la production de l’anticorps murin recherché de type IgG1 et dirigé contre le TNF-α (hybridome HTNF40).
L’humanisation de l’anticorps (12) repose sur le greffage des régions obligatoires courtes et hypervariables (complementary determining region, CDR) de l’anticorps murin sur le fragment d’une immunoglobuline Fab humaine IgGγ1κ.
Les acides aminés de la région charpente (framework, FR) sont également transférés. Les régions CDR d’origine murine constituent le site de liaison de l’antigène, les acides aminés de charpente contribuent au maintien de l’affinité pour l’antigène. L’anticorps monoclonal ainsi obtenu est considéré comme à 98 % humain et conserve l’affinité et la spécificité de l’anticorps murin initial (12, 13).
Seul est conservé le fragment Fab′ humanisé. Celui-ci est couplé chimiquement à deux molécules de polyéthylène glycol de 20 kDa chacune.
Le TNFα soluble présente un poids moléculaire de 17 kDa alors que celui du TNFα membranaire est de 26 kDa (14).
Tableau XI. Renseignements généraux et galéniques du certolizumab (42, 45)
DCI, Spécialité, Laboratoire
Forme galénique, Dosage,
Présentation Excipients
AMM, Liste, Remboursement
Conservation
Certolizumab pegol CIMZIA® UCB
Solution injectable 200 mg/mL Boite 2 seringues pré-remplies de 1 ml avec 2 tampons alcoolisés
Acétate de sodium, chlorure de sodium, eau PPI
* AMM européenne 2009 * Liste I * Médicament soumis à prescription initiale hospitalière annuelle * Prescription initiale et renouvellement réservés aux spécialistes en rhumatologie ou en médecine interne * Médicament d’exception disponible en officine de ville * Agrément à l’usage des collectivités * Inscription sur la liste des spécialités pharma-ceutiques prises en charge en sus des prestations d’hospita-lisation selon l’Arrêté du 9/08/87 en cours.
* Conservation entre +2 et +8 °C * Conserver la seringue pré-remplie dans l’emballage extérieur à l’abri de la lumière.
* Ne pas congeler.
En bref
Le certolizumab pegol est un fragment Fab’ d’anticorps humanisé recombinant, dirigé contre le TNF-α. Le certolizumab se lie au TNF-α avec une grande affinité et le neutralise. L’efficacité du certolizumab pegol administré par voie sous-cutanée a été évaluée dans plusieurs études cliniques. Le certolizumab est indiqué dans le traitement de la PR active, modérée à sévère, chez les malades adultes en association au MTX en cas de réponse inadéquate aux traitements de fond ou en monothérapie en cas d’intolérance au MTX. La posologie est de 400 mg en dose initiale, suivi de 400 mg à S2 et S4 puis 200 mg toutes les 2 semaines. Un plan de gestion des risques a été mis en place pour surveiller les risques infectieux et carcinogènes.

-31-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Figure 2. Représentation des sites spécifiques de Figure 3. Structure comparée des anti-TNFα (13) PEGylation du certolizumab (13) La PEGylation (15) vise à augmenter la demi-vie du fragment Fab′. La demi-vie de l’anticorps monoclonal sous sa forme Ig G a été évaluée chez le rat à 104 heures, alors que celle du fragment Fab′ a été estimée à seulement 22,7 heures. La conjugaison au PEG a permis l’obtention d’une demi-vie de 45,8 heures chez le rat (13). Elle est réalisée sur un site distant du site de liaison au TNF-α. A côté de son action sur la demi-vie, la PEGylation améliore : - la stabilité de la structure moléculaire, - la solubilité en solution aqueuse, - la biodisponibilité.
Le certolizumab pegol se lie au TNF-α avec une grande affinité et le neutralise sélectivement sans neutralisation associée du TGFβ (ou lymphotoxine alpha). Ce dernier critère le différencie de l’etanercept. In vitro, une concentration de 4 ng/mL produit une inhibition 90 % [IC90] (16). Comme l’ont montré des essais sur des systèmes biologiques in vitro, la liaison du certolizumab pegol est dose-dépendante et neutralise à la fois le TNF-α humain soluble et membranaire. Les constantes de dissociation respectives sont : 89,3 pM pour le certolizumab pegol, 157,4 pM pour
l’adalimumab et 227,2 pM pour l’infliximab (13). Dans le cadre d’un essai sur le modèle murin L929 avec des lignées cellulaires humaines, la IC 90 pour le TNF-α soluble a été obtenue à : . 9 ng/mL pour l’infliximab, . 9 ng/mL pour l’adalimumab, . 3 ng/mL pour le certolizumab pegol . 0,7 ng/mL pour l’etanercept (14, 16).
Sur le TNFα membranaire, la puissance d’inhibition des anti-TNF, tels que l’infliximab, l’adalimumab et le certolizumab, a été comparable. L’etanercept est 2 fois moins puissant (14).
Comme la structure du certolizumab pegol ne présente pas de fragment Fc (à la différence de l’infliximab, de l’etanercept et de l’adalimumab), la fixation du complément est rendue impossible. Il s’en suit l’absence in vitro : - de cytotoxicité dépendante des anticorps ou ADCC, - d’apoptose, - de dégranulation des polynucléaires neutrophiles (13, 14, 17).
3.2.2. Données pharmaco-cinétiques
Chez le malade atteint de PR, à l’état d’équilibre :
- le volume de distribution apparent est de 6 à 8 L, - la clairance totale de 21 mL/h - la demi-vie d’élimination terminale de 14 jours environ.
La biodisponibilité du certolizumab pegol après administration par voie sous-cutanée est de 80 à 100 % (13).
3.3. Efficacité clinique
L’efficacité thérapeutique (18, 44) du certolizumab pegol administré par voie sous-cutanée chez des malades atteints de PR active a été évaluée au cours de 3 études randomisées, contrôlées, multicentriques de phase III : - en association au méthotrexate dans les études RAPID 1 (cf. tableau XII) (19) et RAPID 2 (cf. tableau XIII) (20). Ces 2 études ont servi à l’obtention de l’autorisation de mise sur le marché, - en monothérapie et à une posologie différente de celle retenue pour l’AMM, l’étude FAST4WARD (cf. tableau XIV) (21).
Chez les malades atteints de PR, le certolizumab pegol diminue significativement (p < 0,001) les taux de CRP comparativement au placebo quand il est administré en monothérapie ou en association au MTX.

-32-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau XII. Etude RAPID 1 – étude de phase III, randomisée, évaluant l’efficacité et la tolérance de 2 schémas d’administration de certolizumab pegol sous forme lyophilisée en association au méthotrexate dans la polyarthrite rhumatoïde ayant eu une réponse inadéquate au méthotrexate (19)
Etude RAPID 1. Rheumatoid Arthritis PreventIon of structural Damage - Keystone et al, Arthritis and rheumatism – 2008 (19).
Méthodologie
Objectif Evaluation de l’efficacité et de la tolérance de 2 schémas d’administration de certolizumab pegol sous forme lyophilisée par rapport au placebo en association au méthotrexate chez des malades atteints de PR active ayant eu une réponse inadéquate au méthotrexate seul. Type d’étude Etude de phase III, randomisée, contrôlée, en double-aveugle contre placebo, multicentrique. Médicament expérimental : formulation lyophilisée du certolizumab pegol. Schéma et posologie Selon une randomisation 2 :2 :1 3 bras de traitement : Bras 1 : certolizumab pegol 400 mg + méthotrexate (MTX) à S0, S2 et S4 puis 400 mg toutes les 2 semaines Bras 2 : certolizumab pegol 400 mg + MTX à S0, S2 et S4 puis 200 mg toutes les 2 semaines Bras 3 : placebo toutes les 2 semaines + méthotrexate Les malades non répondeurs ACR 20 aux semaines 12 et 14 sortent de l’étude et sont éligibles pour l’étude d’extension ouverte et reçoivent le certolizumab pegol à raison de 400 mg toutes les 2 semaines. Durée : 52 semaines
Inclusion – Evaluation
Inclusion - Malades adultes d’au moins 18 ans, atteints d’une PR ≥ 6 mois et de moins de 15 ans d’évolution, diagnostiquée selon les critères ACR (nombre d’articulations gonflées et nombre d’articulations douloureuses ≥ 9, CRP ≥ 15 mg/L ou VS ≥ 30 mm/h), non contrôlée par le méthotrexate à une dose stable (>10 mg/semaine) depuis au moins 2 mois avant l’inclusion. - Tout DMARD en dehors du méthotrexate, est arrêté 28 jours avant l’initiation ; pour le léflunomide, l’arrêt doit avoir lieu 6 mois avant et être associé à une procédure de wash-out. Exclusion - Toute autre pathologie inflammatoire. - Antécédents de tuberculose, tuberculose latente. - Pathologie démyélinisante. - Malade à haut risque infectieux. - Tumeur. - Toute pathologie sévère active. - Malade ayant reçu une biothérapie dans les 6 mois (etanercept et/ou anakinra dans les 3 mois). - Hypersensibilité, réactions d’anaphylaxie à une biothérapie antérieure - Echec antérieur à un anti-TNF.
Evaluation Co-critères principaux : ACR 20 à 24 semaines, évolution du mTSS* entre l’inclusion et S52. Critères secondaires : HAQ-DI à S24 et S52, ACR 20 à S52, ACR 50 et ACR 70 à S24 et S52.
Résultats Nombre de malades 982 malades randomisés : Bras 1 (400 mg toutes les 2 semaines) : 390 - Bras 2 (Induction 400 mg puis 200 mg) : 393 - Bras 3 (placebo) : 199 Analyse des malades Pourcentage des malades ayant complété l’étude (52 semaines) : Bras 1 : 70,3 % (274) Bras 2 : 64,9 % (255) Bras 3 : 21,6 % (43) Causes des sorties : à S16, 62,8 % des malades du groupe placebo sont sortis de l’étude pour manque d’efficacité, 21,1 % dans le bras 2 et 17,4 % dans le bras 1. Un malade de chaque groupe a été perdu de vue. Profil des malades Les 3 groupes sont comparables Age moyen : 52 ans Pourcentage de femmes : 83,3 % Ancienneté de la maladie (années) : 6,14 ans Nombre de DMARDs (hors méthotrexate) : 1,3 DAS 28 : 7 CRP (mg/L) : 15,3 mg/L HAQ-DI : 1,7
…/…
* Score total de Sharp modifié par Van der Heijde (mTSS) : évaluer la réduction de la progression des dommages structuraux.

-33-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau XII (suite et fin). Etude RAPID 1 – étude de phase III, randomisée, évaluant l’efficacité et la tolérance de 2 schémas d’administration de certolizumab pegol sous forme lyophilisée en association au méthotrexate dans la polyarthrite rhumatoïde ayant eu une réponse inadéquate au méthotrexate (19)
Résultats cliniques (suite)
Efficacité clinique
S24 Bras 1
400 mg toutes les 2 sem.
Bras 2 400 mg à l’induction puis 200 mg toutes les 2 sem.
Contrôle p
ACR 20* 60,8 % 58,8 % 13,6 % < 0,001 ACR 50 39,9 % 37,1 % 7,6 % < 0,001 ACR 70 20,6 % 21,4 % 3,0 % < 0,001
* critère principal
A S52, les résultats d’efficacité clinique se maintiennent significativement. L’action du certolizumab pegol est rapidement obtenue après la première injection. A la semaine 1, la proportion de malades ayant une réponse ACR 20 est plus importante dans les bras 1 et 2 (22,3 % et 22,9 % respectivement) par rapport au placebo (5,6 %) (p<0,001). La réponse ACR 20 optimale est obtenue à S 12. La réponse maximale ACR 50 et ACR 70 dans le bras 2 est obtenue à partir de 14 à 20 semaines de traitement.
Progression radiographique (*) : mTSS à S52 Bras 1 vs contrôle : 0,2 versus 2,8 p < 0,001 Bras 2 versus contrôle : 0,4 versus 2,8 p < 0,001 Le certolizumab pegol en association au méthotrexate inhibe significativement la progression des dommages structuraux par rapport au placebo plus méthotrexate. Aucune différence n’est observée entre les 2 groupes certolizumab pegol au regard de l’inhibition de la progression des érosions et des pincements.
Fonction physique L’amélioration de la fonction physique est significativement plus élevée dans les groupes certolizumab pegol par rapport au placebo, de la semaine 1 à la semaine 52.
Effets indésirables L’exposition au traitement est exprimée en nombre de malade-année : bras 1 : 315,2 ; bras 2 : 303,3 et bras 3 : 91,4. La majorité des effets observés ont été d’intensité moyenne à modérée. La survenue d’infections a conduit 6 malades dans chaque groupe certolizumab pegol, à arrêter le médicament expérimental. Les effets indésirables les plus souvent rencontrés en dehors des infections sont : les maux de tête, l’hypertension, les douleurs lombaires. Une faible incidence des douleurs liées à l’injection et de réactions au site d’injection est observée : respectivement 2 et 2,3 % dans le bras 2, 1,3 et 0,8 % dans le bras 1, aucun signalement dans le groupe placebo. La fréquence des infections est comparable selon les groupes : de 56 à 58 pour 100 malades-année. Les infections les plus fréquentes sont les infections du tractus urinaire et du tractus respiratoire supérieur, les rhinopharyngites. Les infections sévères sont plus nombreuses dans les groupes certolizumab pegol que dans le groupe placebo : 5,3 pour 100 malades-année dans le bras 2, 7,3 dans le groupe 1 contre 2,2 dans le bras placebo. Les infections les plus souvent rencontrées sont celles du tractus urinaire et du tractus respiratoire inférieur, des tuberculoses (5 cas ont été enregistrés dans les groupes traités par certolizumab), des gastroentérites. Des tumeurs ont été observées chez 12 malades. Elles se répartissent : 1 dans le groupe placebo, 7 dans le bras 2, 4 dans le bras 1. Enfin, des anticorps anti-certolizumab pegol ont été détectés à S52 chez 6,4 % des malades ayant reçu le médicament.
Conclusion des auteurs
Cette étude a montré l’efficacité clinique et radiologique d’une association méthotrexate-certolizumab pegol par rapport au placebo plus méthotrexate. Cette efficacité est mise en évidence très rapidement. Le profil de tolérance à court terme semble proche de celui des autres anti-TNF. La PEGylation a permis de minimiser les effets indésirables de type douleur ou réaction au site d’injection.
Conclusion du CNHIM
Une différence statistiquement significative (p <0,001) en terme d’efficacité clinique (ACR 20 à 24 semaines) a été mise en évidence dans le groupe traité par certolizumab pegol avec le schéma posologique retenu dans l’indication AMM (dose initiale de 400 mg à S0, S2 et S4 suivie d’une dose d’entretien de 200 mg toutes les 2 semaines), par rapport au groupe placebo. A 52 semaines, la progression des signes radiologiques a été significativement moindre chez les malades traités par certolizumab pegol par rapport au groupe placebo. Les arrêts de traitement ont été nombreux, en particulier dans le groupe placebo. Bien que le certolizumab pegol soit un fragment d’anticorps, des anticorps anti-certolizumab ont été détectés. Une phase d’extension en ouvert a montré un maintien de l’efficacité du certolizumab pegol sur une durée de 2 ans.

-34-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau XIII. Etude RAPID 2 – étude de phase III, randomisée, évaluant l’efficacité et la tolérance de 2 schémas d’administration de certolizumab pegol (solution injectable) par rapport au placebo, associé au méthotrexate, dans la polyarthrite rhumatoïde active (20)
Etude RAPID 2. Rheumatoid Arthritis PreventIon of structural Damage – 2008 (20).
Méthodologie
Objectif Evaluation de l’efficacité et de la tolérance de 2 schémas d’administration de certolizumab pegol (formulation solution injectable) par rapport au placebo, associé au méthotrexate (MTX), chez des malades atteints de PR active. Type d’étude Etude de phase III, randomisée, contrôlée, en double-aveugle contre placebo, multicentrique et internationale. Médicament expérimental : formulation liquide du certolizumab pegol. Schéma et posologie Selon une randomisation 2 :2 :1 3 bras de traitement : Bras 1 : certolizumab pegol 400 mg + MTX à S0, S2 et S4 puis 400 mg toutes les 2 semaines Bras 2 : certolizumab pegol 400 mg + MTX à S0, S2 et S4 puis 200 mg toutes les 2 semaines Bras 3 : placebo toutes les 2 semaines + méthotrexate Dans le cas d’une absence de réponse ACR 20 à la fois à S 12 et à S 14, le malade est considéré comme un malade non-répondeur selon l’ACR 20. Il peut alors être inclus dans l’étude d’extension en ouvert à S 16 et bénéficier d’un traitement de certolizumab pegol à raison de 400 mg toutes les 2 semaines. Durée : 24 semaines
Inclusion – Evaluation
Inclusion - Malades adultes d’au moins 18 ans avec une PR depuis au moins 6 mois et de moins de 15 ans d’évolution, diagnostiquée selon les critères ACR (nombre d’articulations gonflées et nombre d’articulations douloureuses ≥ 9, CRP ≥ 15 mg/L ou VS ≥ 30 mm/h). - Malades sous méthotrexate depuis au moins 6 mois, à une dose stable ≥ 10 mg/semaine depuis au minimum 2 mois avant l’initiation du médicament expérimental. Exclusion - Tout malade ayant reçu une biothérapie pendant les 6 mois précédant la randomisation (3 mois dans le cas de l’étanercept et de l’anakinra). - Antécédent d’hypersensibilité sévère ou réaction anaphylactique à un agent biologique - Antécédent de tuberculose Evaluation Critère principal : ACR 20 à 24 semaines. Critères secondaires : ACR 50 et ACR 70, différentiel du score total de Sharp modifié par Van der Heijde (TSS) à S 24, fonction physique (HAQ-DI) et qualité de vie (SF-36).
Résultats
Nombre de malades 619 malades randomisés : Bras 1 : 246 - Bras 2 : 246 - Bras 3 : 127 Analyse des malades Pourcentage des malades ayant complété l’étude (24 semaines) : Bras 1 : 73,6 % (181) Bras 2 : 70,7 % (174) Bras 3 : 13,4 % (17) Causes des sorties : manque d’efficacité (en particulier, dans le groupe placebo, 84,3 % des cas), survenue d’effet indésirable (1,6 % pour le bras placebo, 4,5 % pour le bras 2, 2,4 % pour le bras 1). Profil des malades Les 3 groupes sont comparables Age moyen: 51,9 ans Pourcentage de femmes : 82 % Ancienneté de la maladie (années) : 6 ans (de 5,6 dans le bras 3 à 6,5 dans le bras 1) Nombre de DMARDs (hors MTX) : 1,2 CRP (mg/L) : 13,6 HAQ-DI : 1,6 …/…

-35-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau XIII (suite et fin). Etude RAPID 2 – étude de phase III, randomisée, évaluant l’efficacité et la tolérance de 2 schémas d’administration de certolizumab pegol (solution injectable) par rapport au placebo, associé au méthotrexate, dans la polyarthrite rhumatoïde active (20)
Résultats cliniques (suite)
Efficacité clinique
S 24 Bras 1
400 mg toutes les 2 semaines
Bras 2 400 mg à l’induction puis 200 mg toutes les 2 sem.
Contrôle p
ACR 20* 57,6 % 57,3 % 8,7 % < 0,001
ACR 50 33,1 % 32,5 % 3,1 % < 0,001 ACR 70 10,6 % 15,9 % 0,8 % < 0,01
* critère principal
DAS 28 < 2,6 (rémission) à S24 :
Bras 2 vs contrôle : 9,4 % vs 0,8 % p < 0,05 Bras 1 vs contrôle : 8,5 % vs 0,5 % p < 0,05 Critère HAQ-DI (proportion des malades ayant atteint, MCID) : Bras 2 vs contrôle : 57 % vs 11 % p < 0,001 Bras 1 vs contrôle : 53 % vs 11 % p < 0,001 Progression radiographique : Bras 2 vs contrôle : 0,2 vs 1,2 p < 0,01 Bras 1 vs contrôle : -0,4 vs 1,2 p < 0,01 Effets indésirables Ils ont été observés chez 52,8 % des malades du bras placebo, 56 % du bras 2, 50,8 % du bras 1. Les effets indésirables les plus souvent décrits sont : - infections urinaires, hématurie et bactériurie dans le bras placebo, - infections urinaires, infections du tractus respiratoire haut, maux de tête dans le bras 2, - hypertension et maux de tête dans le bras 1. Les effets indésirables ayant conduit à un arrêt du médicament expérimental : 1,6 % dans le groupe placebo, 4,8 % dans le bras 2, 2,8 % dans le bras 1. Des anticorps anti-certolizumab ont été détectés chez 5,1 % des malades. L’incidence des effets indésirables sévères a été de 3,2 % dans le groupe placebo, 7,3 % dans chacun des groupes certolizumab pegol. Parmi les infections sévères, il a été observé des cas : d’érysipèle, des gastroentérites. Cinq cas de tuberculose ont été observés chez des malades sous certolizumab. Un cas de tumeur maligne a été rapporté dans chacun des groupes.
Conclusion des auteurs
Le certolizumab pegol améliore rapidement et significativement les signes et symptômes de la PR. De plus, il inhibe la progression des dommages structuraux, et améliore la fonction physique des malades.
Conclusion du CNHIM
Une différence statistiquement significative a été mise en évidence en faveur du groupe traité par certolizumab pegol à la posologie retenue dans l’AMM (induction à la dose de 400 mg à S0, S2 et S4 puis entretien à la dose de 200 mg toutes les 2 semaines) en association au méthotrexate, par rapport au groupe placebo plus méthotrexate sur la réponse à l’ACR 20 à S 24 (critère principal de l’étude). Une phase d’extension en ouvert est en cours.

-36-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau XIV. Etude FAST4WARD – étude de phase III, randomisée, évaluant l’efficacité et la tolérance du certolizumab pegol 400 mg en monothérapie administré par voie sous-cutanée dans la polyarthrite rhumatoïde active après échec à au moins 1 DMARD, par rapport au placebo (21)
Etude FAST4WARD. EFficAcy ad Safety of CerTolizumab Pegol 4 Weekly dosAge in RheumatoiD Arthritis –2008 (21).
Méthodologie
Objectif Evaluation de l’efficacité et de la tolérance du certolizumab pegol 400 mg en monothérapie administré par voie sous-cutanée chez des malades atteints de PR active après échec à au moins 1 DMARD, par rapport au placebo. Type d’étude Etude de phase III, randomisée, contrôlée, en double-aveugle contre placebo, multicentrique, dans 3 pays (Australie, USA et République de Tchécoslovaquie). Médicament expérimental = forme lyophilisée du certolizumab pegol Schéma et posologie Selon une randomisation 1 : 1 2 bras de traitement : Bras 1 : certolizumab pegol 400 mg toutes les 4 semaines Bras 2 : placebo toutes les 4 semaines Durée : 24 semaines
Inclusion – Evaluation
Inclusion - Malades adultes atteints de PR active selon les critères de l’ACR (nombre d’articulations gonflées et nombre d’articulations doulou-reuses ≥ 9, CRP > 10 mg/L ou VS ≥ 28 mm/h), depuis au moins 6 mois, après manque d’efficacité ou d’intolérance à au moins 1 DMARD (dont l’arrêt a été effectué depuis au moins 28 jours ou depuis 5 demi-vies du médicament concerné ; pour le léflunomide, élimination à l’aide de la colestyramine puis wash-out de 28 jours). Exclusion - Tout rhumatisme inflammatoire autre que la PR. - Toute infection chronique, sévère ou tuberculose. - Tout traitement avec une biothérapie ou traitement antérieur avec anti-TNF pendant les 6 mois précédent la randomisation. Evaluation Critère principal : ACR 20 à 24 semaines. Critères secondaires : ACR 50 et ACR 70, fonction physique (HAQ-DI), SF-36, DAS 28 (VS)3.
Résultats
Nombre de malades 220 malades randomisés : Bras 1 : 111 - Bras 2 : 109 Analyse des malades Pourcentage de malades ayant complété l’étude (24 semaines) : Bras 1 : 68,5 % (76) Bras 2 : 25,7 % (28) Causes des sorties Bras placebo (81) : manque d’efficacité (75), effets indésirables (2), perdu de vue (3), violation au protocole (1) Bras certolizumab pegol (35) : manque d’efficacité (24), effets indésirables (5), perdu de vue (0), violation au protocole (4), retrait de consentement (2) Le pourcentage d’arrêts de traitement a été important dans le groupe placebo, majoritairement par manque d’efficacité. Profil des malades Les 2 groupes sont comparables Age moyen: 53,8 ans Pourcentage de femmes : 83,7 % Ancienneté de la maladie (années) : de 8,7 ans dans le bras 1 à 10,4 dans le bras 2. Nombre de DMARDs antérieur : 2 CRP (mg/L) : 11,5 Score HAQ-DI : 1,5 DAS 28 (VS)3 : 6,3 …/…

-37-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau XIV (suite et fin). Etude FAST4WARD – étude de phase III, randomisée, évaluant l’efficacité et la tolérance du certolizumab pegol 400 mg en monothérapie administré par voie sous-cutanée dans la polyarthrite rhumatoïde active après échec à au moins 1 DMARD, par rapport au placebo (21)
Efficacité clinique (suite)
S 24 Bras 1 Contrôle p ACR 20* 45,5 % 9,3 % < 0,001
ACR 50 22,7 % 3,7 % < 0,001
ACR 70 5,5 % 0 < 0,05 * critère principal
La réponse à l’ACR 20 est significativement en faveur du certolizumab pegol. Amélioration de la fonction physique : HAQ-DI
Bras 1 Contrôle p Semaine 1 -0,23 0,04 <0,001
Semaine 24 -0,36 0,13 <0,001 Effets indésirables (EI) - EI : Bras 1 : 75,7 % - Bras 2 : 57,8 %. La majorité de ces EI ont été d’intensité faible à modérée. Les effets indésirables les plus souvent observés sont : les maux de tête, les rhinopharyngites, les infections du tractus respiratoire supérieur, les diarrhées et les sinusites.
- EI sévères : Bras 1 : 7,2 % (soit 18 cas pour 100 malades-année) - Bras 2 : 2,8 % (soit 9 cas pour 100 malades-année). Aucun cas de tuberculose ou d’infection opportuniste n’a été observé. - Infections sévères : aucun cas dans le bras placebo, 2 cas sous certolizumab pegol. -Douleur au site d’injection : aucun cas de douleur observée dans le bras certolizumab pegol. - Réactions au site d’injection : Bras1 : 4,5 % - Bras 2 : 13,5 % - Auto-anticorps : 8,1 % des malades traités par certolizumab pegol.
Conclusion des auteurs
Une différence statistiquement significative a été mise en évidence en faveur du groupe traité par certolizumab pegol à la posologie de 400 mg toutes les 4 semaines par rapport au groupe traité par placebo sur la réponse à l’ACR 20 à la 24ème semaine, chez des malades en échec de traitements de fond classiques. Comme l’etanercept ou l’adalimumab, le certolizumab pegol peut être utilisé en monothérapie.
Conclusion du CNHIM
Au cours de l’étude, le nombre de malades sortis pour insuffisance d’efficacité a été significativement moins important dans le groupe certolizumab pegol (21, 6 %) par rapport au placebo (68,8 %) (p < 0,001). A partir de S12, les malades, en l’absence de réponse clinique, étaient encouragés à entrer dans l’étude en ouvert. Cette étude met en évidence l’efficacité du certolizumab pegol en monothérapie à la dose de 400 mg toutes les 4 semaines, schéma thérapeutique qui n’a pas été retenu dans l’AMM. Comme l’adalimumab et l’etanercept, le certolizumab pegol peut être prescrit en monothérapie. Toutefois, des études récentes (dont les études Tempo ou Premier) ont montré que les anti-TNF en association au méthotrexate étaient plus efficaces qu’en monothérapie. Qu’en sera-t-il pour le certolizumab pegol ?
3.4. Renseignements théra-peutiques
Cf. tableau XV.
* L’étude Fast4Ward (21) a pu montrer l’efficacité du certolizumab pegol en monothérapie. Toutefois, la spécialité Cimzia est recommandée en première intention en association au méthotrexate, pour deux raisons :
- le taux de répondeurs ACR 20 est plus important avec le certolizumab pegol en association avec le méthotrexate ;
- la proportion d’auto-anticorps est plus importante chez les malades traités en monothérapie.
3.4.1. Posologie et mode d’administration
Le schéma posologique retenu par l’AMM comprend une dose initiale de 400 mg de certolizumab pegol, suivie par deux doses de 400 mg à S2 puis à S4, puis une dose d’entretien de 200 mg tous les deux semaines.
Le contenu total (soit 1 ml) de la seringue pré-remplie doit être
injecté, par voie sous-cutanée. Les sites d’injection adaptés sont la cuisse ou l’abdomen. Après une formation sur la technique d’injection, les malades peuvent s’auto-injecter leur médicament.
3.4.2. Effets indésirables (42)
Cf. tableau XVI. Le certolizumab pegol a été étudié chez 2 367 malades atteints de PR au cours des études contrôlées et ouvertes (phases d’extension), d’une durée maximale de 57 mois.

-38-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau XV. Renseignements thérapeutiques du certolizumab pegol (42)
Spécialité Indication AMM Posologie Contre-indication
CIMZIA®
Traitement de la PR active, modérée à sévère, chez les malades adultes : En association au MTX en cas de : . réponse inadéquate aux traitements de fond (DMARDs), y compris le méthotrexate. En monothérapie en cas d’intolérance au MTX ou lorsque la poursuite du traitement par MTX est inadaptée chez ces malades.*
. Dose initiale de 400 mg . 400 mg à S 2 et S4 . Dose d’entretien de 200 mg toutes les 2 semaines Administration par voie sous-cutanée, au niveau de la cuisse ou de l’abdomen.
. Hypersensibilité à la substance active ou à l’un des excipients. . Tuberculose évolutive ou autres infections sévères telles que sepsis ou infections opportunistes . Insuffisance cardiaque modérée à sévère (NYHA, classes III/IV)
Cas particulier des infections : L’incidence des infections a été plus élevée sous certolizumab pegol par rapport au placebo (0,91 par malade-année contre 0,72). Les infections les plus souvent rencontrées ont été : - les infections des voies respiratoires hautes et basses, - les infections virales à herpès virus, - les infections urinaires.
L’incidence des infections graves a été plus importante dans les bras traités par certolizumab pegol : 0,06 par malade-année contre 0,02 par malade-année pour les groupes placebo. Les infections les plus souvent rencontrées ont été : des cas de tuberculoses (0,5 % contre aucun cas sous placebo), des infections opportunistes invasives de type zona disséminé, pneumocys-tose, œsophagite fongique, ou nocardiose.
Réactions liées à la perfusion : Des réactions au point d’injection ont pu être observées au cours des études cliniques, notamment : des érythèmes, prurits, hématomes, douleurs, gonflements ou ecchymoses. Elles ont concerné 6,4 % des malades traités par certolizumab pegol contre 6,5 % des malades sous placebo. Des douleurs au site d’injection ont été observées chez 1,5 % des malades traités par certolizumab pegol, aucun cas n’ayant nécessité l’arrêt du médicament. Il semblerait que la présence du PEG dans la structure moléculaire du certolizumab pegol inhiberait la dégranulation des mastocytes stimulés. Ces cellules hautement réactives sont capables de sécréter
rapidement des médiateurs de l’inflammation. Aussi, l’effet du PEG sur les mastocytes pourrait expliquer le faible taux de douleur au site d’injection observé sous certolizumab pegol (22).
Immunogénicité : Les malades ayant reçu le certolizumab pegol en association au méthotrexate ont eu un taux de formation d’anticorps plus faible que ceux qui l’ont reçu en monothérapie.
Affections malignes : En dehors des carcinomes cutanés non mélanomes, 30 cancers, y compris 3 cas de lymphomes, ont été observés au cours des études dans la PR (4 136 malades-année).
L’incidence des lymphomes a été estimée à 0,07 pour 100 malades-année, celle des mélanomes à 0,02 pour 100 malades-année.
3.4.3. Mises en garde et précautions d’emploi (42)
Les malades doivent être surveillés étroitement afin de détecter tout signe ou symptôme en relation avec une infection, en particulier la tuberculose.
Le traitement par CIMZIA® ne doit pas être initié chez les malades ayant des infections évolutives, cliniquement importantes, y compris des infections chroniques ou localisées, tant que celles-ci ne sont pas contrôlées.
Comme pour les autres anti-TNF, les malades doivent faire l’objet d’une recherche de tuberculose active ou latente avant traitement. Dans le cas d’une tuberculose latente, un
traitement antituberculeux doit être mis en œuvre. En l’état actuel des connaissances, le risque potentiel de développer des lymphomes ou d’autres tumeurs malignes ne peut être exclu chez les malades traités par anti-TNF. Des précautions doivent donc être prises quand ce type de traitement est envisagé chez un malade ayant des antécédents de cancer.
Le certolizumab pegol est contre-indiqué en cas d’insuffisance cardiaque modérée à sévère. La spécialité CIMZIA® doit être utilisée avec prudence lors d’une insuffisance cardiaque légère (classes I/II).
Comme avec les autres anti-TNF, des troubles hématologiques ont été observés. Il convient des surveiller régulièrement la numération formule sanguine et l’hémogramme.
Comme des infections graves et des neutropénies ont été rapportées au cours des études cliniques lors de l’administration simultanée d’anakinra ou d’abatacept avec l’etanercept (autre anti-TNF), le certolizumab pegol ne doit donc pas être associé à l’anakinra ou l’abatacept.
L’administration de vaccins vivants ou atténués n’est pas recommandée pendant le traitement par Cimzia®.
Il existe une interférence entre le certolizumab pegol et certains tests de la coagulation. Aussi, son utilisation peut entraîner des valeurs de TCA faussement élevées chez des malades sans anomalie de la coagulation.

-39-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau XVI. Effets indésirables du certolizumab (42)
Pathologie / Fréquence Fréquent (> 1/100 et < 1/10) Peu fréquent (>1/1 000 et < 1/100)
Infections et infestations infections bactériennes (incluant abcès), infections virales (herpès virus, papillomavirus, influenza virus)
sepsis, tuberculose, infections fongiques (incluant candidose, histoplasmose, pneumocystose).
Affections du système immunitaire
vascularite, lupus érythémateux, hypersensibilité médicamenteuse, manifestations allergiques, auto-anticorps.
Affection gastro-intestinale ascite, ulcération et perforation gastro-intestinale, inflammation du tractus digestif, stomatite, dyspepsie, distension abdominale, sécheresse oropharyngée.
Affections de l’oreille et du labyrinthe
Vertige.
Affection du système nerveux céphalées (incluant migraine), anomalies sensorielles
neuropathie périphérique, sensations vertigineuses tremblements.
Affections psychiatriques anxiété, troubles de l’humeur.
Affections hépatobiliaires hépatite (incluant l’élévation des enzymes hépatiques)
hépatopathie, cholestase, hyperbilirubinémie.
Paramètres sanguins élévation des phosphatases alcalines sériques, allongement du temps de coagulation
Affections cardiaques cardiomyopathie, coronaropathie ischémique, arythmie, palpitations.
Affections vasculaires hypertension hémorragie ou tout saignement, hypercoagulabilité, syncope, œdème, ecchymoses.
Affections hématologiques et ou du système lymphatique
troubles des éosinophiles, leucopénie (incluant neutropénie, lymphopénie)
anémie, lymphadénopathie, thrombocytopénie, thrombocytose.
Troubles du métabolisme et de la nutrition
déséquilibre hydroélectrolytique, dyslipidémie, troubles de l’appétit, modification du poids
Troubles généraux et anomalies au site d’administration
pyrexie, douleur (toute localisation), asthénie, prurit (toute localisation), réactions au site d’injection
frissons, syndrome pseudo-grippal, altération de la perception de la température, sueurs nocturnes, bouffées vasomotrices.
Affections oculaires troubles visuels, inflammation oculaire et palpébrale, troubles de la sécrétion lacrymale.
Affections de la peau et du tissu sous-cutané
éruptions Alopécie, psoriasis et manifestations apparentées, dermatite et eczéma, affection des glandes sudoripares, ulcère cutané, photosensibilité, acné, décoloration de la peau, sécheresse de la peau, des ongles et affections du lit ungéal.
Affections respiratoires, thoraciques et médiastinales
asthme et syndromes apparentés, épanchement et symptômes pleuraux, congestion et inflammation des voies respiratoires, toux.
Affections musculo-squelettiques et systémiques
troubles musculaires, élévation de la créatine phosphokinase sérique
Affections du rein et des voies urinaires
insuffisance rénale, hématurie, symptômes vésicaux et urétraux.
Affections des organes de reproduction et du sein
troubles du cycle menstruel et saignements utérins, symptômes mammaires.
Tumeurs bénignes, malignes et non précisées (incluant kystes et polypes)
tumeurs solides, carcinomes cutanés hors mélanomes, lésions pré-cancéreuses, tumeurs bénignes et kystes.

-40-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
3.4.4. Grossesse et allai-tement (42)
L’administration de Cimzia® n’est pas recommandée pendant la grossesse. Les femmes susceptibles de procréer doivent utiliser une méthode de contraception efficace et la poursuivre au moins 5 mois après la dernière administration de CIMZIA®. Dans l’état actuel des connaissances, la décision, soit de poursuivre / d’arrêter l’allaitement, soit de poursuivre / d’arrêter le traitement par CIMZIA®, doit être prise en tenant compte du bénéfice de l’allaitement pour l’enfant et du bénéfice du traitement par le certolizumab pegol pour la mère.
3.4.5. Surdosage (42)
Aucune toxicité dose-dépendante n’a été observée au cours des études cliniques. Des doses répétées allant jusqu’à 800 mg par voie sous-cutanée et jusqu’à 20 mg /kg par voie intraveineuse ont été administrées.
3.5. Plan de gestion des risques
Comme tous les autres médicaments de cette classe, la spécialité CIMZIA® fait l’objet d’un plan de gestion des risques qui prévoit : - la surveillance des risques identifiés et potentiels, - en particulier infectieux et carcinogènes. Chaque malade traité reçoit une carte spéciale de surveillance.
4. Golimumab
4.1. Renseignements gé-néraux et galéniques (42)
Cf. tableau XVII.
4.2. Propriétés pharmaco-logiques
4.2.1. Mode d’obtention et mode d’action
Le golimumab (17) est un anticorps monoclonal humain de type IgG1κ produit sur une lignée cellulaire d’hybridome de souris par l’ADN recombinant. A une concentration de 4 ng/mL, le golimumab neutralise in vitro 50 % du TNF-α recombinant humain (23). Il présente une affinité à la fois pour le TNF-α soluble et le TNF-α transmembranaire.
4.2.2. Propriétés pharmaco-cinétiques
Après une administration unique de 50 mg de golimumab à des sujets sains ou malades atteints de PR, le délai médian pour atteindre les concentrations sériques maximales (Tmax) a varié de 2 à 6 jours. La concentration sérique maximale moyenne (Cmax) a été de 3,1 µg/mL.
La pharmacocinétique du golimumab est quasi-linéaire chez les volontaires sains comme chez les malades atteints de PR avec des doses variant de 0,1 à 10 mg/kg administrées en une seule injection intraveineuse.
Après une seule injection sous-cutanée de 100 mg, l’absorption de golimumab a été comparable dans le bras, l’abdomen et les cuisses, avec une biodisponibilité absolue moyenne de 51 % (42).
Chez les malades atteints de PR, recevant une injection sous-cutanée de 50 mg toutes les 4 semaines, le profil pharmacocinétique est le suivant :
- obtention de l’état d’équilibre à la 12ème semaine ; - concentration plasmatique moyen-ne à l’état d’équilibre (Css) égale à 0,4 – 0,6 µg/mL (23).
La clairance apparente a été estimée à 1,91 L/jour pour les malades
atteints de PR. Des variabilités interindividuelles sur la clairance de l’ordre de 35 à 42 % ont été observées selon le poids, les concentrations de la CRP, la présence d’anticorps anti-golimumab ou encore le statut du malade par rapport au tabac (24).
La demi-vie terminale a été estimée à 12 ± 3 jours chez des sujets sains et des valeurs comparables ont été observées chez des malades atteints de PR, de rhumatisme psoriasique ou de spondylarthrite ankylosante (24, 42).
Aucun ajustement de la posologie de golimumab n’est recommandé en fonction de l’âge, ou du sexe. De même, il n’existe pas de recom-mandation particulière concernant son utilisation chez les malades insuffisants rénaux et/ou hépatiques (23).
4.3. Efficacité clinique
L’évaluation clinique du golimumab dans la PR repose sur 3 études cliniques : - l’étude GO-AFTER (cf. tableau XVIII) (25), après échec à un ou plusieurs anti-TNF ; - l’étude GO-BEFORE (cf. tableau XIX) (26), malades naïfs de MTX ; - l’étude GO-FORWARD (cf. tableau XX) (27), malades naïfs d’anti-TNF, en cas d’échec au MTX.
Dans ces études, ont été évalués pour : - GO-AFTER : 461 malades ayant été précédemment traités avec un ou plusieurs anti-TNF, l’adalimumab, l’etanercept ou l’infliximab ; - GO-FORWARD a évalué 444 malades atteints de PR active malgré un traitement au méthotrexate, à une dose stable d’au moins 15 mg/semaine et n’ayant jamais reçu d’anti-TNF.
Pour chacune d’elle, le critère principal retenu était le pourcentage de malades ayant obtenu une réponse à l’ACR 20 à la semaine 14. L’étude GO-FORWARD a évalué également l’amélioration du score du HAQ-DI à S24.
4.4. Renseignements théra-peutiques (42)
Cf. tableau XXII.
En bref
Le golimumab est un anticorps monoclonal humain de type IgG1 produit sur une lignée cellulaire d’hybridome de souris par ADN recombinant. L’efficacité clinique du golimumab a été évaluée par plusieurs études cliniques. Le golimumab est indiqué dans le traitement de la PR active modérée à sévère en association avec le MTX en cas de réponse inadéquate aux traitements de fond. La posologie est de 50 mg en SC une fois par mois.

-41-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau XVII. Renseignements généraux et galéniques du golimumab (42)
DCI, Spécialité Laboratoire
Forme galénique Dosage,
Présentation
Excipients
AMM Liste
Collectivité Remboursement
Conservation
Golimumab
SIMPONI®
Schering plough
Solution injectable en stylo prérempli (SmartJect) Un stylo de 0,5 ml contient 50 mg de golimumab
Sorbitol (E420), L-histidine, chlorhydrate d’histidine mono-hydraté, polysorbate 80, eau PPI
AMM Européenne EU/1/09/546/001
Non inscrit sur la liste des spécialités
remboursables à la date du 2 octobre 2010
. Entre +2 et +8°C
. Ne pas congeler
. Conserver dans l’emballage extérieur à l’abri de la lumière
Tableau XXII. Renseignements thérapeutiques du golimumab (42)
Indications AMM européenne
Posologie Contre-indications
PR active modérée à sévère de l’adulte, en association avec le MTX lorsque la réponse aux traitements de fond classiques, y compris le MTX, a été inadéquate.
50 mg par voie sous-cutanée, une fois par mois. Administration de manière concomi-tante avec le méthotrexate.
- Hypersensibilité à la substance active ou à l’un des excipients. - Tuberculose active ou autres infections sévères, telles que sepsis et infections opportunistes. - Insuffisance cardiaque modérée ou sévère (de classe III/IV dans la classification NYHA)
4.4.1. Mode d’administration (42)
Le golimumab est proposé à la commercialisation sous une forme injectable destinée à la voie sous-cutanée. Après une formation préalable, les malades pourront s’injecter eux-mêmes leur médicament.
L’injection de SIMPONI® doit être effectuée une fois par mois, à la même date chaque mois. En cas d’oubli d’une injection, le malade ne doit surtout pas injecter une double dose pour compenser la dose oubliée mais doit appliquer le principe suivant : - si la dose est administrée avec moins de deux semaines de retard, le malade devra s’injecter la dose oubliée et poursuivre selon le calendrier mensuel initial ;
- si la dose est administrée avec plus de deux semaines de retard, le malade devra s’injecter la dose oubliée et un nouveau calendrier mensuel devra être établi à partir de la date de cette dernière injection. Comme pour les autres anti-TNF administrés par vois sous-cutanée, le médicament s’injecte :
- généralement, sur le devant du milieu des cuisses ; - en-dessous de l’estomac (abdomen) sous le nombril, sauf dans la zone de 5 cm directement en-dessous du nombril. Il ne faut pas injecter le médicament au niveau de zones où la peau présente un hématome, des vergetures ou une cicatrice, ainsi que si celle-ci est rouge, squameuse ou rigide.
La spécialité SIMPONI® sera proposée sous forme d’un stylo prérempli à usage unique appelé SMARTJECT®. Après avoir retiré le stylo prérempli du réfrigérateur, celui-ci doit être maintenu à température ambiante pendant 30 minutes avant l’injection du golimumab. Le stylo ne doit pas être secoué.
4.4.2. Effets indésirables (24, 42) Cf. tableau XXIII. Les données de tolérance sont issues des essais cliniques menés dans les 3 indications retenues dans l’AMM européenne : la PR, le rhumatisme psoriasique et la spondylarthrite ankylosante. Cela représente un total de 2 578 malades dont 1 600 atteints de PR.

-42-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau XVIII. Etude GO-AFTER – étude de phase III, randomisée, évaluant l’efficacité et la tolérance de golimumab dans la polyarthrite rhumatoïde active, précédemment traitée par un ou plusieurs anti-TNF (25)
Etude GO-AFTER. GOlimumab After Former anti-tumour necrosis factor αααα Therapy Evaluated in Rheumatoid arthritis – 2009 (25)
Méthodologie Objectif Evaluer l’efficacité et la tolérance de golimumab chez des malades atteints de PR active et qui ont reçu précédemment un ou plusieurs anti-TNF.
Type d’étude Etude de phase III randomisée, contrôlée, en double aveugle et multicentrique.
Schéma et posologie 3 bras de traitement : randomi-sation selon le ratio 1 :1 :1
BRAS 1 : une injection de placebo (seringue de 0,5 ml) et une injection de placebo (seringue de 1 ml) par voie sous-cutanée, toutes les 4 semaines BRAS 2 : une injection de golimumab 50 mg (seringue 0,5 ml) et une injection de placebo (seringue de 1 ml) par voie sous-cutanée, toutes les 4 semaines BRAS 3 : une injection de golimumab 100 mg (seringue de 1 ml) et une injection de placebo (seringue de 0,5 ml) par voie sous-cutanée, toutes les 4 semaines.
L’utilisation concomitante de méthotrexate, sulfasalazine ou hydroxychloroquine, seuls ou associés est autorisée.
Stratification selon le site de l’étude et l’utilisation du méthotrexate.
Traitement de secours possible à S16 pour les malades du groupe placebo ou golimumab 50 mg dans le cas d’une absence de réponse ACR 20 %.
Durée : 24 semaines
* Natalizumab, autorisé en France dans la prise en charge de la sclérose en plaques (TYSABRI®) et dans la Maladie de Crohn aux USA. ** Efalizumab, n’est plus autorisé en Europe (cas de LEMP, rapport bénéfice/risque défavorable), dans la prise en charge du psoriasis (RAPTIVA®), reste commercialisé aux USA. Il s’agit d’un anticorps anti-CD11a. *** Alefacept, commercialisé aux USA, indiqué dans la prise en charge du psoriasis (AMEVIVE®). Ce médicament interfère avec l’activation des lymphocytes T.
Inclusion – Evaluation Inclusion - Malades âgés de 18 ans et plus atteints de PR≥ 3 mois - Nombre d’articulations gonflées ≥ 4 et d’articulations douloureuses ≥ 4. - Les malades doivent avoir reçu au moins un anti-TNF α avant l’inclusion. - La dernière dose d’anti-TNF α a été faite avant la première administration du médicament expérimental : 8 semaines avant pour l’adalimumab ou l’etanercept, 12 semaines avant pour l’infliximab.
Exclusion - Toute pathologie inflammatoire autre que la PR, - Toute réaction sévère à un anti-TNF antérieur, - Tout malade ayant reçu : . le natalizumab* ou le rituximab, . l’anakinra moins de 4 semaines avant, . l’alefacept**ou l’efalizumab*** moins de 3 mois avant l’administration de la première dose du médicament expérimental. . des médicaments cytotoxiques - Toute infection granulomateuse latente ou active, toute infection opportuniste moins de 6 mois avant l’inclusion, toute infection sévère ou chronique, - Toute pathologie démyélinisante, -Toute pathologie associée non contrôlée, sévère. Evaluation Critère principal : ACR 20 à 14 semaines. Critères secondaires : ACR 20 à S 24, ACR 50 et ACR 70 à S 14 et S 24, DAS 28 à S 14 et S 24, score HAQ-DI à S 14 et S 24, FACIT-F à S 14 et S 24
Résultats Nombre de malades 461 malades randomisés : Bras 1 : 155 (dont 72 ont bénéficié d’un traitement de secours de golimumab 50mg à S16) - Bras 2 : 153 (dont 41 ont bénéficié d’un traitement de secours de golimumab 100mg à S16) – Bras 3 : 153 Analyse des malades Malades arrivant au bout de l’étude (S24) : Bras 1 : 124 (dont 69 avec le traitement de secours de golimumab 50mg à S16) soit 80 % Bras 2 : 140 (dont 38 avec le traitement de secours de golimumab 100 mg à S16) soit 91,5 % Bras 3 : 138 soit 90 % Causes de sortie de l’étude : Bras 1 : 10 cas d’EI (dont 1 concerne les malades du traitement de secours), 11 cas pour manque d’efficacité (dont 2 concernent les malades du traitement de secours), 1 cas de décès, 9 autres cas, cause non connue. Bras 2 : 4 cas d’EI, 6 cas pour manque d’efficacité (dont 3 concernent les malades du traitement de secours), 2 autres cas cause non connue. Bras 3 : 2 cas d’EI, 5 cas pour manque d’efficacité, 1 cas de décès, 2 perdus de vue, 2 autres cas cause non connue. Profil des malades Les 3 groupes sont comparables Age moyen: 54,6 ans Pourcentage de femmes : 79,6 Ancienneté de la pathologie : 9,3 ans DAS 28 : 6,2 HAQ-DI : 1,6 CRP : 8,6 mg/L Environ deux tiers des malades de chaque groupe sont sous méthotrexate. …/…

-43-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau XVIII (suite et fin). Etude GO-AFTER – etude de phase III, randomisée, évaluant l’efficacité et la tolérance de golimumab dans la polyarthrite rhumatoïde active, précédemment traitée par un ou plusieurs anti-TNF (25)
Résultats cliniques
Efficacité clinique (suite) * Semaine 14
placebo golimumab 50 mg golimumab 100 mg ACR 20 18 % 35 % p=0,0006 $ 38 % p=0,0001 $ ACR 50 6 % 16 % p=0,0062 $ 20 % p=0,0003 $ ACR 70 2 % 10 % p=0,0018 $ 9 % p=0,0050 $
DAS 28 placebo golimumab 50 mg golimumab 100 mg < 2,6* 1 % 8 % p=0,0009 $ 12 % p<0,0001 $
< 3,2 15 % 23 % p=0,0001 34 % p=0,0003 $ 3,2-5,1 46 % 49 % p=0,5778 43 % p=0,6316 > 5,1 39 % 28 % p=0,0529 23 % p=0,0036
(* DAS 28 < 2,6 = rémission ; $ = résultats significatifs)
Réponse EULAR : . Bras 1 (placebo) : 27 % . Bras 2 (golimumab 50 mg) : 49 % p = 0,0001 . Bras 3 (golimumab 100 mg) : 34 % p < 0,0001 HAQ- DI : . Bras 1 (placebo) : 0 % . Bras 2 (golimumab 50 mg) : - 13 ,4 % p = 0,0005 . Bras 3 (golimumab 100 mg) : -17,6 % p < 0,0001 FACIT-F (score) : . Bras 1 (placebo) : 1 . Bras 2 (golimumab 50 mg) : 6 p = 0,0004 . Bras 3 (golimumab 100 mg) : 5 p = 0,0005
* Semaine 24
placebo golimumab 50 mg golimumab 100 mg
ACR 20 17 % 34 % p=0,0005 $ 44 % p<0,0001 $ ACR 50 5 % 18 % p=0,0003 $ 20 % p=0,0001 $ ACR 70 3 % 12 % p=0,0041 $ 10 % p=0,0107 $
DAS 28 placebo golimumab 50 mg golimumab 100 mg < 2,6* 3 % 10% p=0,0049 $ 16 % p=0,0001 $ < 3,2 14 % 31 % p=0,0010 37 % p<0,0001 $ 3,2-5,1 37 % 39 % p=0,7924 47 % p=0,1083
> 5,1 49 % 30 % p=0,0019 16 % p<0,0001 (* DAS 28 < 2,6 = rémission ; $ = résultats significatifs)
Réponse EULAR : Bras 1 (placebo) : 25 % Bras 2 (golimumab 50 mg) : 46 % p = 0,0001 Bras 3 (golimumab 100 mg) : 61 % p < 0,0001 HAQ – DI : Bras 1 (placebo) : 0 % Bras 2 ( golimumab 50 mg) : - 13,4 % p = 0,0003 Bras 3 (golimumab 100 mg) : - 14,3 % P < 0,0001 FACIT-F (score) : Bras 1 (placebo) : 3 Bras 2 (golimumab 50 mg) : 5 p = 0,0211 Bras 3 (golimumab 100 mg) : 6 p = 0,0003
Effets indésirables . EI : bras 1 (placebo) 70 %, bras 2 (golimumab 50 mg) : 61 %, bras 3 (golimumab 100 mg) : 73 %. . EI sévères : bras 1 (placebo) 7 %, bras 2 (golimumab 50 mg) : 5 %, bras 3 (golimumab 100 mg) : 3 %. . Infections : bras 1 (placebo) 28 %, bras 2 (golimumab 50 mg) : 27 %, bras 3 (golimumab 100 mg) : 25 %. . Aucun cas de tuberculose observé. . Réaction au point d’injection : bras 1 (placebo) 3 %, bras 2 (golimumab 50 mg) : 4 %, bras 3 (golimumab 100 mg) : 11 %. . Effets indésirables les plus fréquents : infections du tractus respiratoire supérieur, rhinopharyngites, diarrhées, arthralgies, sinusites, hypertension, toux.
Conclusion des auteurs
Ces résultats montrent que le golimumab pourrait être une alternative aux anti-TNF dans le traitement de la PR en cas d’échec aux DMARDs et autres anti-TNFα.
Conclusion du CNHIM
Cette étude a été menée sur des effectifs moyens. Il convient de souligner que 72 malades sur les 155 randomisés dans le bras placebo ont eu besoin d’un traitement de secours à S16. Ces résultats montrent l’efficacité clinique et l’amélioration de la fonction physique pour les malades traités par golimumab par rapport au placebo mais ils ne permettent pas de départager le golimumab 50 mg et le golimumab 100 mg. L’un des critères principaux d’évaluation est la réponse ACR à S14, donc à un stade plus précoce, contrairement à la plupart des études qui utilisent plutôt l’ACR à S24.

-44-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau XIX. Etude GO-BEFORE – étude de phase III, randomisée, évaluant l’efficacité et la tolérance de golimumab associé au méthotrexate dans la polyarthrite rhumatoïde active (26)
Etude GO-BEFORE. GOlimumab, Before Employing Methotrexate as theFirst-Line Option in the Treatment of Rheumatoid Arthritis of Early Onset - 2009 (26).
Méthodologie Objectif Evaluer l’efficacité et la tolérance de golimumab associé au méthotrexate chez des malades naïfs de méthotrexate, atteints de PR active. Type d’étude Etude de phase III randomisée, contrôlée, en double aveugle et multicentrique. Schéma et posologie 4 bras de traitement Bras 1 : placebo + méthotrexate Bras 2 : golimumab 100 mg + placebo du méthotrexate Bras 3 : golimumab 50 mg + méthotrexate Bras 4 : golimumab 100 mg + méthotrexate Le golimumab est administré par voie sous-cutanée à S0 et toutes les 4 semaines. Le méthotrexate est débuté à la dose de 10 mg / semaine à S0 puis est augmenté par palier de 2,5 mg / semaine jusqu’à 20 mg / semaine à S8. Durée : 52 semaines Extension en ouvert de 5 ans
Inclusion – Evaluation Inclusion - Malades atteints de PR ≥ 3 mois - PR active, nombre d’articulations gonflées et douloureuses ≥ 4 ; avec au moins deux des critères suivants : CRP ≥ 15 mg/L ou VS ≥ 28 mm/h ; Raideur matinale > 30 min ; érosions osseuses radio-logiques et/ou par IRM ; positivité des anti-CCP ou facteur rhumatoïde. - Malades ayant reçu l’anakinra, si la dernière dose a été faite plus de 4 semaines avant l’administration du médicament expérimental. - Malades susceptibles d’avoir reçu du méthotrexate mais au maximum pendant 3 semaines. Exclusion - Tout malade ayant reçu antérieurement : infliximab, eta-nercept, adalimumab, rituximab, natalizumab ou des médicaments cytotoxiques (chlorambucil, cyclo-phosphamide, agents alkylant, dérivés de la moutarde à l’azote. Evaluation Critère principal : ACR 50 à S 24, variation du score de Sharp modifié par van Heijde à S52. Critères secondaires : ACR 20 ACR 50 et ACR 90, DAS 28, score HAQ-DI
Résultats Nombre de malades 637 malades randomisés : Bras 1 : 160 - Bras 2 : 159 - Bras 3 : 159 - Bras 4 : 159 634 malades traités Bras 1 : 160 – Bras 2 : 157 – Bras 3 : 158 – Bras 4 : 159 Analyse des malades Malades ayant complété l’étude jusqu’à S24 : Bras 1 : 150 soit 94 % Bras 2 : 148 soit 93 % Bras 3 : 150 soit 94 % Bras 4 : 151 soit 95% Causes de sortie de l’étude : Bras 1 : EI (1), manque d’efficacité (1), perdue de vue (3), autres (5) Bras 2 : EI (1), manque d’efficacité (3), autres (5) Bras 3 : EI (5), décès (1), perdue de vue (1), autres (1) Bras 4 : EI (6), décès (1), perdue de vue (1), autres (1) Profil des malades Les 4 groupes sont comparables Age moyen : 49,5 ans Pourcentage de femmes : 82,9 % Ancienneté de la pathologie : 3,5 ans CRP (mg/dL) : 2,5 DAS 28 : 5,1 HAQ-DI : 1,5
…/…

-45-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau XIX (suite et fin). Etude GO-BEFORE – étude de phase III, randomisée, évaluant l’efficacité et la tolérance de golimumab associé au méthotrexate dans la polyarthrite rhumatoïde active (26)
Résultats cliniques (suite)
Bras S 24
Placebo + MTX Golimumab 100 mg + placebo
Golimumab 50 mg + MTX
Golimumab 100 mg + MTX
ACR 50 29,4 % 32,7 % 40,3 % (0,042) 36,5 % (0,177) ACR 20 49,4 % 51,6 % 61,6 % (0,028) 61,6 % (0,028) ACR 70 15,6 % 13,8 % 23,9 % (0,064) 18,2 % (0,535) ACR 90 3,1 % 3,1 % 9,4 % (0,021) 7,5 % (0,080)
Bras
DAS28-CRP Placebo + MTX
Golimumab 100 mg + placebo
Golimumab 50 mg + MTX
Golimumab 100 mg + MTX
Réponse bonne ou modérée
60,6 % 66 % 75,5 % (0,005) 75,5 % (0,004)
Rémission 28,1 % 25,2 % 38,4 % (0,05) 37,7 % (0,069)
Index HAQ – DI : Bras placebo + MTX : 36,95 % Bras golimumab 100 mg + placebo : 31,05 % Bras golimumab 50 mg + MTX : 43,65 % p = 0,141 Bras golimumab 100 mg + MTX : 48,55 % p = 0,006 Amélioration de la fonction physique obtenue sous golimumab plus MTX. Effets indésirables S 24 . Pourcentage de malade ayant développé au moins un EI Bras 1 : 72,5 % Bras 2 : 68,2 % Bras 3 : 81,6 % Bras 4 : 76,1 % Les effets indésirables les plus fréquemment observés dans les groupes golimumab + MTX ont été : nausées, infections du tractus respiratoire supérieur, augmentation du taux des ASAT et des ALAT, dyspepsie, maux de tête. . EI sévères : Bras 1 : 6,9 % Bras 2 : 3,2 % Bras 3 : 6,3 % Bras 4 : 6,3 % . Deux décès pendant la période de 24 semaines : un décès dans le groupe 3 par suicide, un dans le groupe 4. . Réactions locales (le plus souvent érythème) au point d’injection : Bras 1 : 1,9 % Bras 2 : 10,8 % Bras 3 : 4,4 % Bras 4 : 8,8 % . Infections : Bras 1 : 32,5 % Bras 2 : 35 % Bras 3 : 34,2 % Bras 4 : 31,4 % . Infections sévères : Bras 1 : 1,9 % Bras 2 : 1,3 % Bras 3 : 1,3 % Bras 4 : 4,4 % . Détection des anticorps anti-golimumab : chez 13,5% des malades du bras 2, 3,7% des malades du bras 3 et 1,9 % des malades du bras 4. Les malades ayant reçu du golimumab 100 mg seul semblent avoir plus de risque de développer des anticorps que lorsque celui-ci est administré en association au méthotrexate (bras 3 et 4).
Conclusion des auteurs
Cette étude a montré que le golimumab utilisé en association avec le MTX est plus efficace que le MTX utilisé seul chez le malade naïf atteint de PR.
Conclusion du CNHIM
Ces résultats semblent modestes mais le critère principal retenu pour l’évaluation (ACR 50) représente un objectif de réponse plus difficile à atteindre que l’ACR 20 (plus souvent choisi dans les études).

-46-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau XX. Etude GO-FORDWARD – étude de phase III, randomisée, évaluant l’efficacité et la tolérance de golimumab chez des malades atteints de PR active malgré un traitement au méthotrexate (27)
Etude GO-FORDWARD. Golimumab,a human antibody to tumour necrosis factor given by monthly subcutaneous injections, in active rheumatoid arthritis despite methotrexate
therapy – 2009 (27)
Méthodologie Objectif Evaluer l’efficacité et la tolérance de golimumab chez des malades atteints de PR active malgré un traitement au méthotrexate. Type d’étude Etude de phase III randomisée, contrôlée, en double aveugle et multicentrique, internationale. Schéma et posologie - Période de « run-in » pendant laquelle les malades reçoivent du méthotrexate à une dose stable (15 mg/semaine en moyenne). - Randomisation selon le ratio 3 :3 :2 :2 Bras 1 : placebo + méthotrexate Bras 2 : golimumab 100 mg + placebo du méthotrexate Bras 3 : golimumab 50 mg + méthotrexate Bras 4 : golimumab 100 mg + méthotrexate Golimumab : une injection par voie sous-cutanée toutes les 4 semaines. A la semaine 16, les malades des groupes 1, 2, 3 n’ayant pas atteints la réponse ACR 20, changent de thérapeutique : le groupe 1 reçoit le golimumab 50 mg plus méthotrexate, le groupe 2 reçoit du méthotrexate à la place du placebo plus golimumab 100 mg, le groupe 3 reçoit golimumab 100 mg à la place du golimumab 50 mg plus méthotrexate. Durée : 52 semaines Extension en ouvert de 5 ans.
Inclusion – Evaluation Inclusion -Malades atteints de PR ≥ 3 mois - PR active, avec un nombre d’articulations gonflées et douloureuses ≥ 4. - Malades ayant au moins deux des critères suivants : CRP ≥ 15 mg/L ou VS ≥ 28 mm/h, raideur matinale > 30 min, érosions osseuses radiologiques et/ou par IRM ; positivité des anti-CCP ou facteur rhumatoïde. - Malades sous méthotrexate à dose stable (15 mg/semaine, au maximum 25 mg/semaine) pendant les 4 semaines précédant la sélection. Exclusion - Tout malade ayant reçu antérieurement : infliximab, etanercept, adalimumab, rituximab, natalizumab ou des médicaments cytotoxiques, anakinra, DMARD autres que le méthotrexate. Evaluation Critère principal : ACR 20 à S14, amélioration de la fonction physique HAQ-DI à S 24 Critères secondaires : ACR 50/70, DAS 28
Résultats Nombre de malades 444 malades randomisés : Bras 1 : 133 – bras 2 : 133 – Bras 3 : 89 – bras 4 : 89. Analyse des malades Malades ayant complété l’étude jusqu’à S24, sans changement de traitement à S16 : Bras 1 : 84 soit 70,6 % Bras 2 : 91 soit 68,4 % Bras 3 : 72 soit 80,9 % Bras 4 : 82 soit 92,1 % Causes de sortie de l’étude : Bras 1 : EI (4), manque d’efficacité (1), perdue de vue (1), autres (1) Bras 2 : EI (5), décès (1), arrêt traitement oral (1) Bras 3 : EI (2) Bras 4 : EI (5), perdue de vue (1), arrêt traitement oral (1) - Changement de traitement à S16 : Bras 1 : 41 malades dont 39 complètent l’étude jusqu’à S24 Bras 2 : 36 malades dont 34 complètent l’étude jusqu’à S24 Bras 3 : 15 malades dont 15 complètent l’étude jusqu’à S24. Bras 4 : 0 Profil des malades Les 4 groupes sont comparables Age moyen : 51,3 ans Pourcentage de femmes : 80,7 % Ancienneté de la pathologie : 5,9 ans CRP (mg/L) : 0,9 DAS 28 (CRP) : 4,9 HAQ-DI : 1,3 …/…

-47-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau XX (suite et fin). Etude GO-FORDWARD – étude de phase III, randomisée, évaluant l’efficacité et la tolérance de golimumab chez des malades atteints de PR active malgré un traitement au méthotrexate (27)
Résultats cliniques (suite) : En ITT
Bras S 14
Placebo + MTX Golimumab 100 mg +
placebo Golimumab 50 mg +
MTX Golimumab 100 mg +
MTX ACR 20* 33,1 % 44,4 % (0,059) 55,1 % (0,001) 56,2% (<0,001) ACR 50 9,8 % 20,3 % (0,016) 34,8 % (<0,001) 29,2 % (<0,001)
ACR 70 3,8 % 7,5 % (0,184) 13,5 % (0,008) 9 % (0,104) * critère principal
Amélioration de l’index HAQ – DI par rapport à la valeur initiale (S 24)* : Bras placebo + MTX : -0,13 Bras golimumab 100 mg + placebo : -0,13 (0,240) Bras golimumab 50 mg + MTX : -0,38 (<0,001) Bras golimumab 100 mg + MTX : -0,50 (<0,001) Amélioration de la fonction physique obtenue sous golimumab plus MTX.
Bras S 24
Placebo + MTX
Golimumab 100 mg + placebo
Golimumab 50 mg + MTX
Golimumab 100 mg + MTX
ACR 20 27,8 % 35,3 % (0,187) 59,6% (<0,001) 59,6 % (<0,001) ACR 50 13,5 % 19,5 % (0,187) 37,1 % (<0,001) 32,6 % (<0,001) ACR 70 5,3 % 11,3 % (0,075) 20,2 % (<0,001) 14,6 % (0,017)
Effets indésirables
Avant S16 . Pourcentage de malades ayant développé au moins un EI : Bras 1 : 60,9 % Bras 2 : 63,2 % Bras 3 : 68,5 % Bras 4 : 69,7 % . Pourcentage de malades ayant développé au moins un EI sévère : Bras 1 : 2,3 % Bras 2 : 3,8 % Bras 3 : 5,6 % Bras 4 : 9 % . Pourcentage de malade ayant développé au moins une réaction au point d’injection : Bras 1 : 2,3 % Bras 2 : 3 % Bras 3 : 4,5 % Bras 4 : 4,5 % . Pourcentage de malades ayant développé au moins une infection : Bras 1 : 24,1 % Bras 2 : 30,1 % Bras 3 : 28,1 % Bras 4 : 28,1 % . Pourcentage de malades ayant développé au moins une infection sévère : Bras 1 : 0,8 % Bras 2 : 0,8 % Bras 3 : 2,2 % Bras 4 : 5,6 % . 1 cas de tumeur dans le bras 4.
S 24 Effets indésirables sévères : Bras 1 : 3,7 % (0,09 événement par malade-année) Bras 2 : 6 % (0,11 évènement par malade-année) Bras 3 : 4,2 % (0,04 événement par malade-année) Bras 4 : 12,4 % (0,18 événement par malade-année)
Infections sévères : Bras 1 : 0,7 % (0,02 événement par malade-année) Bras 2 : 3 % (0,05 évènement par malade-année) Bras 3 : 0,9 % (0,02 événement par malade-année) Bras 4 : 4,8 % (0,08 événement par malade-année)
Réactions au site d’injection : Bras 1 : 3 % Bras 2 : 7,5 % Bras 3 : 2,4 % Bras 4 : 4,8 %
. Tumeurs : Au total, 4 malades (1 dans le bras 1, 2 dans le bras 2 et 1 dans le bras 4).
Conclusion des auteurs
L’association golimumab plus méthotrexate a montré une amélioration significative des signes et des symptômes de la PR, ainsi que de la fonction physique. Le groupe golimumab 100 mg plus MTX est associé à une incidence beaucoup plus élevée de survenue d’infection, un malade est décédé d’une infection sévère.
Conclusion du CNHIM
Le schéma de cette étude est relativement complexe. Les résultats sont en faveur du bras golimumab 50 mg plus méthotrexate, qui offre le meilleur rapport bénéfice/risque. Ces résultats restent toutefois à confirmer, une étude d’extension en ouvert est en cours.
Les résultats concernant les effets du golimumab sur la progression radiologique, issus des études GO-BEFORE (malades naïfs de méthotrexate) et GO-FORWARD (après réponse inadéquate au méthotrexate) ont été présentés lors du dernier Congrès de l’ACR. A un an, les premières conclusions font état pour le malade :
- naïf de méthotrexate, d’une inhibition significative démontrée dans les bras golimumab 50 mg plus méthotrexate et golimumab 100 mg plus méthotrexate par rapport au méthotrexate seul ;
- en échec de méthotrexate, d’absence de résultats significatifs. Ces études font l’objet d’extensions à 5 ans.

-48-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau XXI. Etude de phase III, randomisée, évaluant l’efficacité et la tolérance du golimumab administré par voie intraveineuse dans la polyarthrite rhumatoïde (28)
Golimumab, a New Human Anti-Tumor Necrosis Factor α Antibody, Administered Intravenously in Patients With Active Rheumatoid Arthritis – 2010 (28)
Méthodologie Objectif Evaluer l’efficacité et la tolérance de golimumab administré par voie intraveineuse chez les malades atteints de PR Type d’étude Etude de phase III randomisée, en double aveugle, contrôlée par un placebo, multicentrique et internationale. Schéma et posologie 5 bras de traitement selon le ratio : 1 :1 :1 :1 :1 Bras 1 : Perfusion intraveineuse de placebo + méthotrexate Bras 2 : Perfusion de golimumab 2 mg/kg + méthotrexate Bras 3 : Perfusion de golimumab 2mg/kg + placebo du méthotrexate Bras 4 : Perfusion de golimumab 4 mg/kg + méthotrexate Bras 5 : Perfusion de golimumab 4 mg/kg + placebo du méthotrexate Perfusion intraveineuse sur 30 minutes, toutes les 12 semaines. Durée : 48 semaines
Inclusion – Evaluation Inclusion - Malades adultes atteints de PR selon les critères ACR, active en dépit d’un traitement de méthotrexate (15 – 25 mg par semaine) depuis au moins 3 mois avant inclusion, à un dosage stable depuis au moins 4 semaines. - Nombre d’articulations gonflées et douloureuses ≥ 4 - CRP ≥ 15 mg/L et une VS ≥ 28 mm /h - Raideur matinale ≥ 30 min. - Une érosion osseuse mise en évidence par radiographie ou IRM - Détection positive de Facteur rhumatoïde ou anti-CCP Exclusion - Tout malade ayant reçu : . le natalizumab, l’abatacept ou le rituximab, . l’anakinra moins de 4 semaines avant la première dose du médicament expérimental, . l’alefacept, l’efalizumab ou l’infliximab moins de 3 mois avant l’administration de la première dose du médicament expérimental, . l’adalimumab ou l’etanercept moins de 2 mois avant l’administration de la première dose du médicament expérimental. Evaluation Critère principal : ACR 50 à S 14, HAQ-DI et la variation de la CRP, Critères secondaires : ACR 50 à S 24, ACR 20 à S 14, DAS 28- CRP à S 14 et le score SF-36 à S 14.
Résultats Nombre de malades 643 malades randomisés : Bras 1 : 129, Bras 2 : 129, Bras 3 : 128, Bras 4 : 128, Bras 5 : 129. 514 malades traités avec golimumab, avec ou sans méthotrexate 129 malades traités avec placebo plus méthotrexate. Analyse des malades Nombre de malades ayant complété l’étude : difficultés de lecture de l’analyse. Profil des malades Les groupes sont comparables Age moyen: 49,5 ans Pourcentage de femmes : 81 % Ancienneté de la pathologie : 8,1 ans CRP (mg/L) : 18
…/…

-49-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau XXI (suite et fin). Etude de phase III, randomisée, évaluant l’efficacité et la tolérance du golimumab administré par voie intraveineuse dans la polyarthrite rhumatoïde (28)
Résultats cliniques (suite)
Efficacité clinique (* critère principal - ** critère secondaire)
placebo golimumab S 14
placebo + MTX 2 mg/kg + MTX 2 mg/kg + placebo
4 mg/kg + MTX 4 mg/kg + placebo
ACR50* 13,2 % 21,7 % (0,073) 12,5 % (0,872) 21,1 % (0,093) 19,4 % (0,175) ACR20** 27,9 % 55 % (<0,001) 39,8 % (0,043) 51,6% (<0,001) 48,1% (<0,001) ACR 70 4,7 % 7 % (0,420) 3,9 % (0,769) 5,5 % (0,763) 4,7 % (0,987)
placebo golimumab S 24
placebo + MTX 2 mg/kg + MTX 2 mg/kg + placebo 4 mg/kg + MTX 4 mg/kg + placebo
ACR50** 9,3 % 18,6 % (0,032) 8,6 % (0,844) 25 % (<0,001) 11,6 % (0,540) ACR 20 24,8 % 37,2 % (0,032) 22,7 % (0,687) 50 % (<0,001) 29,5% (0,406) ACR 70 3,1 % 6,2 % (0,240) 3,1 % (0,990) 7,8 % (0,097) 6,2 % (0,236)
placebo golimumab DAS 28-CRP Placebo + MTX 2 mg/kg + MTX 2 mg/kg + placebo 4 mg/kg + MTX 4 mg/kg + placebo S 14 44,2 % 69 % (<0,001) 62,5 % (0,003) 73,4% (<0,001) 64,3% (0,001) S 24 40,3 % 53,5 % (0,035) 37,5 % (0,646) 68 % (<0,001) 48,8 % (0,173)
placebo golimumab Rémission
placebo + MTX 2 mg/kg + MTX 2 mg/kg + placebo
4 mg/kg + MTX 4 mg/kg + placebo
S 14 10 % 11,6 % (0,691) 18 % (0,069) 25 % (0,002) 12,4 % (0,560) S 24 7 % 13,2 % (0,100) 6,3 % (0,816) 24,2 % (<0,001) 10,9 % (0,272)
Effets indésirables
A S 16 : Malades ayant développé au moins 1 EI : .Bras placebo : 67,4 % .Bras golimumab 2 mg/kg : 64,3 % .Bras golimumab 4 mg/kg : 61,5 % .Bras golimumab 2mg/kg + MTX : 68 % .Bras golimumab 4 mg/kg + MTX : 70,6 % Malades ayant développé au moins 1 effet indésirable sévère : .Bras placebo : 1,6 % .Bras golimumab 2 mg/kg : 6,2 % .Bras golimumab 4 mg/kg : 1,5 % .Bras golimumab 2mg/kg + MTX : 3,9 % .Bras golimumab 4 mg/kg + MTX: 4 % Malades ayant développé au moins une infection : .Bras placebo : 33, 3 % .Bras golimumab 2 mg/kg : 29,5 % .Bras golimumab 4 mg/kg : 30,8 % .Bras golimumab 2mg/kg + MTX : 30,5 % .Bras golimumab 4 mg/kg + MTX : 40,5 % Malades ayant développé au moins une infection sévère : .Bras placebo : 0,8 % .Bras golimumab 2 mg/kg : 4,7 % .Bras golimumab 4 mg/kg : aucun .Bras golimumab 2mg/kg + MTX : 1,6 % .Bras golimumab 4 mg/kg + MTX : 1,6 % Nombre de réactions par rapport au nombre de perfusions : .Bras placebo : 7/252 .Bras golimumab 2 mg/kg : 9/252 .Bras golimumab 4 mg/kg : 3/257 .Bras golimumab 2mg/kg + MTX : 7/254 .Bras golimumab 4 mg/kg + MTX : 3/251
A S 48 : 82 % des malades traités par golimumab et 72 % des malades sous placebo associé au méthotrexate ont développé des effets indésirables. Ces résultats montrent qu’il n’existe pas de relation entre le taux global des effets indésirables et la dose de golimumab ou le traitement associé au méthotrexate. Dans les groupes golimumab, les infections les plus fréquemment décrites ont été : les infections du tractus respiratoire supérieur, bronchites, rhinopharyngites, infections urinaires, sinusites. La proportion de malades ayant développé des effets indésirables sévères est plus importante dans les groupes golimumab par rapport au groupe placebo. L’incidence des réactions observées au cours des perfusions est la moins élevée dans le groupe golimumab 4 mg/kg plus méthotrexate, et la plus élevée dans le groupe golimumab 2 mg/kg. Détection des anticorps anti-golimumab : 27 malades sur 529 malades traités soit 5 %. La proportion de malades avec anticorps anti-golimumab est plus importante chez les malades ayant reçu le golimumab en monothérapie (9 %) par rapport à ceux l’ayant reçu en association avec le méthotrexate (3%).
Conclusion des auteurs
L’administration intraveineuse de golimumab en association avec le méthotrexate apporte un bénéfice à long terme dans la réduction des signes et symptômes liés à la PR chez des malades en échec de méthotrexate avec un profil de tolérance acceptable. L’objectif principal à S 14 n’a pas été atteint (p=0,051).
Conclusion du CNHIM
La voie d’administration intraveineuse du golimumab dans cette étude n’a pas été retenue dans l’AMM Européenne.

-50-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau XXIII. Effets indésirables du golimumab (42)
Fréquence Pathologie
Très fréquent (> 1/10)
Fréquent (> 1/100 et < 1/10)
Peu fréquent (> 1/1 000 et < 1/100)
Infections et infestations
Infection des voies respiratoires hautes (naso-pharyngite, pharyngite, laryngite et rhinite)
Infections bactériennes (telle que cellulite), infections virales (telles que grippe et herpès), bronchite, sinusite, infections fongiques superficielles
Infections sévères : choc septique, sepsis, tuberculose Infection des voies respiratoires basses telle que pneumonie Infections opportunistes fongiques invasives (histoplasmose, coccidïomycoses, pneumocystose) Infection bactérienne, mycobactérienne atypique et protozoaire), pyélonéphrite, abcès, arthrite bactérienne, bursite infectieuse
Affections du système immunitaire
Réactions allergiques (bronchospasme, hypersensibilité, urticaire), auto-anticorps positifs
Affection gastro-intestinale
Constipation, dyspepsie, douleur gastro-intestinale et abdominale
Affection du système nerveux
Vertiges, paresthésies, céphalées
Troubles démyélinisants, troubles de l’équilibre, dysgueusie
Affections hépatobiliaires
Augmentation ASAT/ALAT Cholélithiase, troubles hépatiques
Affections cardiaques Insuffisance cardiaque congestive : apparition de novo ou aggravation, arythmie, troubles ischémiques des artères coronaires
Affections vasculaires Hypertension Thrombose, syndrome Raynaud, rougeur
Affections hématologiques et ou du système lymphatique
Anémie Leucopénie, thrombocytopénie
Troubles du métabolisme et de la nutrition
Augmentation du taux de glucose dans le sang, augmentation des lipides
Affections endocriniennes
Trouble thyroïdien (tels que hypo-, hyper-thyroïdie et goitre)
Troubles généraux et anomalies au site d’administration
Pyrexie, asthénie, réaction au site d’injection (telle qu’érythème au site d’injection, urticaire, induration, douleur, hématome, prurit, irritation et paresthésies), gêne thoracique
Affections oculaires Troubles visuels (tels que vision floue et diminution de l’acuité visuelle), conjonctivite, allergie oculaire (tels que prurit et irritation)
Affections psychiatriques
Dépression, insomnie
Affections de la peau et du tissu sous-cutané
Alopécie, dermatite, prurit, rash
Affections rénales et urinaires
Troubles de la vessie
Affections respiratoires, thoraciques et médiastinales
Asthme et symptômes associés (tels que sifflements et hyperactivité bronchique)
Tumeurs bénignes, malignes et non précisées (incluant kystes et polypes)
Tumeurs (telles que cancer de la peau, carcinome à cellules squameuses et naevus

-51-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Infections Lors des études contrôlées de phase III dans le traitement de la PR, du rhumatisme psoriasique (RP) et de la spondylarthrite ankylosante (SA) (autres indications du golimumab) jusqu’à 16 semaines, des infections ont été observées chez 28,3 % des malades traités par golimumab contre 24,7 % chez les malades du groupe contrôle. L’infection des voies respiratoires hautes a été l’effet indésirable le plus fréquent, survenant chez 7,2 % des malades traités par golimumab contre 5,8 % chez les malades du groupe contrôle. Des infections sévères ont été observées chez 1,4 % des malades traités par golimumab et 1,3 % chez les malades du groupe contrôle. Elles comprenaient des cas de tuberculose, d’infections bactériennes y compris sepsis et pneumonie, infections fongiques invasives et autres infections opportunistes. Certaines de ces infections ont été mortelles. Tumeurs malignes - Lymphome Au cours des phases contrôlées des essais cliniques de phase IIb et de phase III, l’incidence des lymphomes chez les malades traités par golimumab atteints de PR, de RP et de SA au cours d’une année de suivi, a été supérieure à celle attendue dans la population générale. Un lymphome a été diagnostiqué chez 2 malades. - Tumeurs malignes autres que le lymphome Dans ces mêmes études, l’incidence des tumeurs malignes autres que le lymphome (excepté les cancers de la peau autres que le mélanome) était comparable dans les groupes golimumab et dans le groupe contrôle. Un cancer de la peau autre que le mélanome a été diagnostiqué chez 19 sujets (5 dans le groupe placebo, 6 dans le groupe golimumab 50 mg et 8 dans le groupe golimumab 100 mg) avec une incidence (IC de 95 %) par 100 sujets-année de suivi de 0,72 (0,39, 1,20) évènement pour le golimumab et 1,51 (0,49, 3,52) évènement pour le groupe placebo. A côté des tumeurs déjà citées, d’autres ont été diagnostiquées chez 12 sujets (2 dans le groupe placebo, 6 dans le groupe golimumab 50 mg et 4 dans le groupe golimumab 100 mg) avec une incidence (IC de
95 %) pour 100 sujets-année de suivi de 0,51 (0,24, 0,94) évènement pour golimumab et 0,60 (0,07, 2,17) évènement pour le groupe placebo. Augmentation du taux des enzymes hépatiques Lors des études sur la PR et la SA jusqu’à la semaine 16, l’augmen-tation du taux d’ALAT supérieure ou égale à 5 fois les limites supérieures à la normale (LNS) était peu fréquente et observée chez un nombre plus important de malades traités par golimumab (0,4% à 0,9%) par rapport aux malades du groupe contrôle (0,0 %). Au cours d’une année de suivi, ces augmentations ont été en général asymptomatiques et les anomalies ont diminué ou se sont résolues soit en maintenant, soit en interrompant le traitement par golimumab, soit en modifiant la prise de médicaments concomitants. Au cours des études de phase II et de phase III sur la PR, le RP et la SA, un malade présentant des anomalies hépatiques préexistantes et recevant des traitements pouvant y être associés, a reçu golimumab et a développé une hépatite fatale non infectieuse accompagnée d’une jaunisse. Le rôle de golimumab comme facteur associé ou aggravant ne peut pas être exclu. Réactions au site d’injection Des réactions au site d’injection ont été observées lors des études contrôlées de phase III de 16 semaines sur la PR, le RP et la SA. Elles sont survenues chez 5,8 % des malades traités par golimumab par rapport à 2,2 % chez les malades du groupe contrôle. Il est possible que la présence d’anticorps anti-golimumab puisse augmenter le risque de réactions au site d’injection. La majorité des réactions au site d’injection ont été légères ou modérées, et la manifestation la plus fréquente a été un érythème au site d’injection. Les réactions au site d’injection n’ont généralement pas nécessité l’arrêt du traitement. Au cours de ces mêmes études, aucun malade traité par golimumab n’a développé de réaction anaphylactique. Auto-anticorps Lors des études de phase III menées dans la PR, le RP et la SA au cours d’une année de suivi, 4,0 % des malades traités par golimumab
et 2,6 % des malades du groupe contrôle ont développé des anticorps antinucléaires (à des titres de 1/160 ou supérieurs).
4.4.3. Mises en garde et précautions d’emploi (42) Les malades traités par golimumab doivent faire l’objet d’une surveillance attentive au regard des infections possibles, y compris la tuberculose. Avant la mise sous traitement, il convient d’éliminer tout risque de tuberculose active ou latente. Dans le cas d’une recherche positive de tuberculose latente, un traitement antituberculeux doit être initié. Le golimumab ne doit pas être administré à des malades atteints d’une infection active et cliniquement importante. Des précautions doivent être prises lors de son utilisation chez des malades présentant une infection chronique ou des antécédents d’infection récurrente. Le rôle potentiel d’un traitement anti-TNF, en particulier le golimumab, dans le développement des tumeurs malignes est inconnu. Le golimumab doit être utilisé avec précaution chez les malades atteints d’insuffisance cardiaque légère (classe I/II de la NYHA). Comme avec les autres anti-TNF, des troubles hématologiques ont été observés. Il convient des surveiller régulièrement la numération formule sanguine et l’hémogramme. En raison d’un risque important de survenue d’infections graves, il n’est pas recommandé d’associer le golimumab à : - l’anakinra ; - l’abatacept. L’administration de vaccins vivants ou atténués n’est pas recommandée pendant le traitement par golimumab.
4.4.4. Grossesse et allai-tement (42) Il n’existe pas de données suffisamment pertinentes concernant l’utilisation du golimumab chez la femme enceinte. A ce titre, son utilisation ne doit pas être recommandée. Une grossesse ne pourra être envisagée qu’après un délai de 6 mois à compter de l’arrêt du golimumab. Les mêmes

-52-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
précautions sont à prendre par rapport à l’allaitement.
4.4.5. Surdosage (42) Des doses uniques allant jusqu’à 10 mg/kg ont été administrées par voie intraveineuse dans le cadre d’une étude clinique sans appa-rition de toxicité limitant la dose. Dans le cas où un surdosage se produirait, il est recommandé de surveiller attentivement le malade afin de détecter tout signe ou symptôme d’effets indésirables et d’initier immédiatement un traite-ment symptomatique approprié.
5. Stratégie thérapeu-tique dans la PR 5.1. Biothérapies de la PR : synthèse des indications validées par une AMM 5.1.1. Anti-TNF Cf. tableau XXIV
5.1.2. Autres classes de biothérapies Cf. tableau XXV
5.2. Proposition de stratégie d’utilisation des biothérapies La prise en charge thérapeutique de la polyarthrite rhumatoïde (PR) a considérablement évolué au cours des dix dernières années, avec la mise sur le marché de médicaments biologiques, dotés d’un effet symptomatique mais également d’une capacité à inhiber la progression de l’atteinte structurale. La disponibilité de ces biothérapies, parallèlement au développement d’outils d’évaluation, a permis de mieux définir les modalités de prise en charge de cette maladie. Notre proposition de stratégie thérapeutique (figure 4) repose sur les référentiels déjà disponibles (recommandations de la Haute Autorité en Santé, de l’Eular et autres recommandations professionnelles données par la Société Française de Rhumatologie (SFR), le CRI) (29-34, 43-44) ainsi que sur l’expérience déjà acquise.
Le traitement de fond de la PR est à instaurer idéalement dans les 3 premiers mois d’évolution de la maladie, et parfois avant même que le diagnostic ne soit définitivement établi. Il est admis qu’un retard de l’ordre de 3 mois est préjudiciable à moyen et long terme tant au niveau du retentissement fonctionnel que de la progression structurale (31). Le choix du traitement de fond initial est guidé par de multiples paramètres en particulier l’activité de la maladie, l’existence d’érosions, la présence de facteur rhumatoïde ou d’anticorps anti-CCP, de manifestations extra-articulaires, de comorbidités et les traitements associés. Classiquement, le traitement de fond de référence est le méthotrexate (33), instauré le plus souvent en monothérapie. Dans les formes les plus bénignes de la PR, des alternatives au méthotrexate existent tels que le léflunomide, la sulfasalazine, l’azathioprine et l’hydroxychloroquine. Dans les formes les plus sévères de la maladie, une biothérapie anti-TNFα peut être instaurée en première intention, en association au méthotrexate. Il s’agit notamment des formes poly-articulaires d’emblée, avec présence érosions précoces et auto-anticorps (facteur rhumatoïde et/ou anticorps anti-CCP). Pour chaque traitement, un délai d’action optimal est défini. A l’issue de cette période, l’efficacité doit être évaluée en vue d’éventuelles adaptations. L’échec relatif au méthotrexate peut conduire selon les cas soit : - à augmenter la dose hebdomadaire jusqu’à 0,3 mg/kg,
- au changement de traitement de fond conventionnel,
- à l’association de traitements de fond conventionnels,
- ou à l’ajout d’une biothérapie notamment anti-TNFα (32).
L’échec à un traitement se définit par l’observation d’un des critères suivants : - la persistance d’une activité clinique et biologique de la PR,
- une progression structurale, qui contraste parfois avec l’obtention d’une réponse clinique satisfaisante,
- la nécessité de maintenir une dose trop élevée de corticoïde (au long cours, la posologie moyenne quotidienne des corticoïdes ne doit pas dépasser 0,10 à 0,15 mg/kg/jour).
Les associations de traitements de fond conventionnels les plus souvent utilisées sont : méthotrexate-hydro-xychloroquine, méthotrexate-sulfasa-lazine et méthotrexate-sulfasalazine-hydroxychloroquine. Les options thérapeutiques après échec au méthotrexate, reposent essentiellement sur les biothérapies. L’instauration d’une biothérapie intervient dans la majorité des cas après échec au méthotrexate ou d’un autre traitement de fond conven-tionnel utilisé à dose optimale ou maximale tolérée. Les médicaments pouvant être prescrits dans ce contexte sont les 3 anti-TNFα actuellement disponibles (infliximab, etanercept, adalimumab), l’anti-récepteur IL-6, le tocilizumab et l’anti-IL-1, l’anakinra. Ce dernier a pratiquement été abandonné en raison de son efficacité insuffisante et de son rythme d’administration jugé trop contraignant : une injection sous-cutanée quotidienne. Les données actuellement publiées ne permettent pas de différencier en termes d’efficacité les 3 anti-TNFα disponibles. En cas d’intolérance ou de contre-indication au méthotrexate, d’autres traitements de fond conventionnels peuvent être associés à l’anti-TNFα, tels que le léflunomide, l’azathioprine ou la sulfasalazine. L’etanercept, l’adalimumab et le tocilizumab peuvent également être prescrits en monothérapie. L’efficacité des anti-TNFα et du tocilizumab se révèle le plus souvent en quelques semaines chez les répondeurs, mais un délai minimal de 3 mois est obligatoire pour s’assurer de l’absence de réponse au traitement. La dose d’etanercept n’est pas modulable, contrairement à celle de l’adalimumab, dont les injections peuvent être administrées à une fréquence hebdomadaire, ou de l’infliximab, pour lequel les perfusions peuvent être rapprochées avec un intervalle de 6 semaines et/ou la dose augmentée de 3 à 5 mg/kg (42).

-53-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Tableau XXIV. Synthèse des indications AMM des anti-TNFα (42-44)
Libellé de l’indication REMICADE® infliximab
ENBREL® etanercept
HUMIRA® adalimumab
CIMZIA® certolizumab
pegol
SIMPONI® golimumab
En association au méthotrexate
PR modérément à sévèrement active, lorsque la réponse aux traitements de fond, y compris le MTX, est inadéquate.
� � � �
PR active, lorsque la réponse aux traitements de fond, y compris le MTX, est inadéquate.
�
PR sévère, active, et évolutive chez des malades non traités précédemment par MTX.
� � �
En monothérapie, si intolérance au MTX ou si la poursuite du MTX est inadaptée
PR modérément à sévèrement active, lorsque la réponse aux traitements de fond, y compris le MTX, est inadéquate.
� � �
PR sévère, active et évolutive chez des malades non précédemment traités par MTX,
� �
Tableau XXV. Synthèse des indications AMM des autres biothérapies (42-44)
Cibles thérapeutiques IL-1 Lymphocytes B Lymphocytes T Récepteur de l’IL-6
Libellé de l’indication KINERET® anakinra
MABTHERA® rituximab
ORENCIA® abatacept
ROACTEMRA® tocilizumab
En association au méthotrexate
Traitement des signes et des symptômes de la PR chez les malades dont la réponse au MTX seul n’est pas satisfaisante
+
PR sévère, active, dans le cas d’une réponse inadéquate ou une intolérance aux traitements de fond, dont au moins un anti-TNF
+
PR modérée à sévère, active, en cas de réponse inadéquate à au moins un traitement de fond classique dont MTX
+ (*) +
PR modérée à sévère, active, en cas de réponse inadéquate à au moins un traitement de fond (anti-TNF).
+ +
En monothérapie, si intolérance au MTX ou si la poursuite du MTX est inadaptée
PR modérée à sévère, active, en cas de réponse inadéquate à au moins un traitement de fond (classique dont MTX ou 1 anti-TNF).
+
(*) Nouvelle indication AMM – Juillet 2010

-54-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Après échec d’un traitement par un premier anti-TNFα, plusieurs options sont possibles comme : - l’utilisation d’un deuxième anti-TNFα,
- ou le recours à un médicament d’une autre classe thérapeutique : l’abatacept, le rituximab ou le tocilizumab.
La perte progressive de l’efficacité du premier anti-TNFα après l’obtention d’une réponse initiale caractérise l’échappement thérapeutique, et peut faire envisager l’utilisation d’un deuxième anti-TNFα (34). En revanche, l’absence de réponse thérapeutique initiale au premier anti-TNFα ou échec primaire conduit préférentiellement à choisir un médicament d’une autre classe : l’abatacept, le rituximab ou le tocilizumab. Le recours à un troisième anti-TNFα après l’échec de deux premiers est reconnu comme moins efficace et n’est pas recommandé (34). Le tocilizumab comme l’abatacept peuvent être prescrits en première ligne de biothérapie, en association au méthotrexate, dans la prise en charge de la PR modérée à sévère active en cas de réponse inadéquate à au moins un traitement de fond classique. Le rituximab n’intervient théoriquement qu’après l’échec à au moins un anti-TNFα. Dans les situations d’intolérance ou d’inadéquation à la poursuite du méthotrexate, seul le tocilizumab peut être alors prescrit en monothérapie. Le rituximab et l’abatacept sont prescrits en association au méthotrexate ou en cas d’impossibilité, à un autre traitement de fond classique.
Le certolizumab pegol, nouvel anti-TNFα, constitue une alternative aux autres médicaments de la même classe disponibles dans la prise en charge de la PR. Mais, aucune étude n’est à ce jour disponible le comparant aux autres anti-TNFα ou ayant évalué son efficacité après échec à un autre anti-TNFα.
Aucune recommandation consen-suelle n’existe actuellement quant à l’utilisation séquentielle de ces agents biologiques. Il est essentiel d’évaluer régulièrement leur efficacité, afin de pouvoir modifier rapidement le traitement en cas de réponse clinique jugée insuffisante ou de progression des lésions anatomiques de la PR.
Deux communications présentées au dernier Congrès de l’Eular illustrent bien l’intérêt des facteurs prédictifs pour le choix thérapeutique. La première concerne les facteurs prédictifs de réponse au rituximab dans la PR avec une réponse inadéquate ou une intolérance aux anti-TNFα. Deux facteurs prédictifs ont été mis significativement en évidence à partir des données extraites d’une étude randomisée en ouvert, l’étude SMART : - la détection positive du facteur rhumatoïde et/ou anticorps anti-CCP (p = 0,002) ;
- un taux d’Ig G supérieur à la limite supérieure de la normale (> 12,7 g/L) (p = 0,043) (35).
La deuxième est relative aux facteurs prédictifs d’intolérance au rituximab. Les données ont été extraites de l’utilisation du rituximab dans la « vraie vie » à partir du Registre AIR mis en place par la Société Française de Rhumatologie. Il en ressort que : - un taux d’Ig G < 6g /L expose le malade à un risque d’infections sévères (p = 0,005) ;
- une pathologie chronique respiratoire et/ou insuffisance cardiaque est défavorable (p = 0,01) (36).
6. Conclusion La polyarthrite rhumatoïde (PR) est le plus fréquent des rhumatismes inflammatoires chroniques, affectant de 180 à 300 000 personnes en France. Cette maladie chronique est grave et invalidante. A ce titre, l’amélioration de la prise en charge des malades atteints de PR et de leur qualité de vie demeure un besoin de santé publique.
En dix ans, les connaissances sur cette pathologie ont nettement progressé, de la confirmation du diagnostic, plus précoce, à la mise en place du traitement le plus approprié.
L’origine est plurifactorielle associant de nombreux facteurs prédisposants potentiels, particulièrement d’ordre hormonal, immunologique, environ-nemental ou psychologique. La PR est une maladie auto-immune dont le déclenchement et la pérennisation font intervenir un dérèglement de l’immunité avec formation d’auto-anticorps. La meilleure connaissance de la physiopathologie a permis la distinction de thérapeutiques
ciblées, visant des cytokines, des cellules immunitaires, lymphocytes T ou lymphocytes B. En parallèle, des outils d’évaluation (évaluation clinique et biologique de l’activité de la maladie) ont été mis à disposition afin de mieux définir les modalités de prise en charge de la maladie. L’objectif principal de la prise en charge thérapeutique est de contrôler l’activité de la maladie et si possible d’induire la rémission, la réduction de la douleur, la préven-tion et le contrôle des destructions articulaires et de prévenir la perte de fonction dans les activités de la vie quotidienne et au travail. Le traitement de la PR repose sur des traitements symptomatiques (visant les poussées, la douleur, l’inflammation locale) et des traitements de fond.
Les traitements de fond (37) ou DMARDs (terminologie anglo-saxonne pour Disease-Modifying Anti Rheumatic Drugs) comprennent : - les traitements de fond conven-tionnels ou « classiques » : notam-ment, le méthotrexate (MTX), la référence, et la sulfasalazine ou le léflunomide. - les premières biothérapies avaient pour cibles thérapeutiques le TNF-α et l’IL-1.
Les anti-TNF-α utilisés en clinique étaient alors au nombre de 3 : l’adalimumab, l’étanercept et l’infliximab. L’utilisation de ces médicaments s’est progressivement affinée. Leur profil de tolérance est maintenant mieux cerné.
L’anakinra est le seul antagoniste du récepteur de IL-1 prescrit. Alors que l’efficacité thérapeutique est satis-faisante, le mode d’administration en une injection quotidienne par voie sous-cutanée, est plus contrai-gnant.
La deuxième génération des biothérapies plus récente concerne des substances actives qui visent les cellules immunitaires : le rituximab (lymphocytes B) et l’abatacept (lymphocytes T). Enfin, deux nouveaux anti-TNF-α sont maintenant proposés : le certolizumab pegol tout récem-ment commercialisé en France, et le golimumab qui a obtenu une AMM Européenne mais qui n’est pas encore disponible en France.

-55-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Figure ??. (a) - dans le cas d’un échappement thérapeutique, plutôt qu’après un échec primaire
Stratégies guidées par les critères suivants :
1 La (les) contre-indications(s) spécifique(s) à une molécule ou à une classe thérapeutique
2 La possibilité de prescrire en monothérapie (*) si contre-indication ou intolérance préalable aux traitements de fond conventionnels (méthotrexate, leflunomide, sulfasalazine)
3 Le choix du malade (par rapport à son vécu, son mode de vie, sa vie professionnelle, etc…)
4 L’observance du malade
5 La présence de facteurs prédictifs d’une non-réponse et/ou d’une intolérance
6 Les recommandations de l’HAS et des sociétés savantes, la détermination de facteurs prédictifs d’intolérance ou d’efficacité
Figure 4. Proposition de stratégie d’utilisation des biothérapies.
Anti-TNFαααα Tocilizumab (*) Abatacept
infliximab etanercept * adalimumab *
certolizumab pegol * golimumab
2ème ligne de biothérapie
2ème anti-TNFαααα Rituximab Tocilizumab Abatacept
(a)
1ère ligne de biothérapie
Cas d’une insuffisance de réponse au méthotrexate à une dose optimale
Cas d’une insuffisance de réponse au 1er anti-TNFαααα
Prochainement
……
association ou non au méthotrexate

-56-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
Dans le précédent Dossier du CHNIM 2010 XXXI-4 ont été développées les monographies du rituximab, anticorps monoclonal dirigé contre le CD 20 et celle de l’abatacept, modulateur sélectif de la co-stimulation des LT.
Le rituximab s’intègre en deuxième ligne de biothérapie dans le cas de PR sévère active associée à une réponse inadéquate ou une intolérance aux traitements de fond, dont au moins un anti-TNF.
L’abatacept, initialement identifié en deuxième ligne de biothérapie, a confirmé récemment son intérêt potentiel, en association au métho-trexate, en première ligne de biothérapie en cas de réponse ina-déquate à au moins un traitement de fond classique dont le métho-trexate. Pour ces deux médicaments, un plan de gestion des risques et des études observationnelles sont mis en place. Leur but est de collecter les données de tolérance à long terme.
Dans la deuxième partie de cet article, Dossier 2010 XXXI-5, trois nouvelles biothérapies sont abordées : le tocilizumab dont la cible est l’IL-6 et plus précisément ses récepteurs (anti-récepteur à l’IL-6), le certolizumab pegol et le golimumab, deux nouveaux anti-TNF-α. Le certolizumab pegol se distingue des autres anti-TNF par sa structure sous forme de fragment Fab’ d’anticorps humanisé recombinant.
Dans la stratégie thérapeutique, le tocilizumab peut être prescrit en première ligne ou deuxième ligne de biothérapie, en association au méthotrexate mais également en monothérapie en cas d’intolérance au méthotrexate et lorsque la poursuite du traitement par MTX est inadaptée chez ces malades.
Le certolizumab pegol possède une AMM en première ligne de biothérapie comme les autres anti-TNF-α, préférentiellement en association au méthotrexate. Aucune étude comparative n’est à ce jour disponible le comparant aux autres anti-TNF-α. Les mêmes contre-indications du certolizumab pegol sont identiques à celles des autres anti-TNF. Une meilleure connaissance des données de tolérance à long terme est maintenant nécessaire.
Enfin, le golimumab pourrait s’intégrer prochainement dans la stratégie thérapeutique comme les autres anti-TNF-α. La commer-cialisation en France n’est pas effective à ce jour dans l’attente de données complémentaires sur son évaluation clinique.
Une troisième génération de biothérapies est en développement dans la PR. Des cytokines, telles que l’IL-17 et l’IL-7, constituent des cibles thérapeutiques potentielles en cours d’évaluation dans la PR (38, 39). Le denosumab, anticorps mono-clonal totalement humanisé dirigé contre le Rank Ligand (action sur la résorption osseuse), est aussi étudié dans la PR (40). L’ocrélizumab et l’ofatumumab, anticorps monoclonaux anti-CD20 totalement humanisés, sont en cours d’étude. L’ofatumumab lui est dirigé contre un épitope différent de la molécule CD20 (41).
La prise en charge globale du malade atteint de PR est complexe et repose sur l’implication de tous les acteurs de santé.
La multiplicité des traitements disponibles, leurs modalités d’administration variables (per os, injectable pour perfusion intravei-neuse ou pour administration sous-cutanée) et leurs effets indésirables potentiels avec la priorité de prévenir les complications éven-tuelles, nécessitent la mise en place de programmes d’éducation thérapeutique du malade.
L’objectif est d’accompagner le malade dans l’acceptation de son état, l’orga-nisation de ses soins et ses comportements de santé afin d’améliorer ou de stabiliser sa qualité de vie.
Le malade doit s’impliquer dans les actions thérapeutiques le concernant (notamment choix du mode d’admi-nistration du médicament par rapport à son mode de vie, sa vie professionnelle ; pour les femmes, anticipation des modifications de traitement lors d’un désir de grossesse).
Aujourd’hui, les biothérapies permettent au plus grand nombre d’atteindre l’état de rémission et contribuent nettement à l’amélioration de la qualité de vie des malades atteints de PR.
Abréviations et acronymes
ACR : American College of Rheumatology.
Anti-CCP : Anticorps antipeptides cycliques citrullinés.
CRP : protéine C activée.
DAS : Disease Activity Score.
DMARD : Disease Modifying Anti-Rheumatic Drug.
EULAR : European League Against Rheumatism.
EVA : échelle visuelle analogique.
FACIT : Functional Assessment of Chronic Illness Therapy.
FR : facteur rhumatoïde.
HAQ-DI : Health Assessment Ques-tionnaire Disability Index.
Ig : immunoglobuline.
IgG : immunoglobuline de type G.
IgM : immunoglobuline de type M.
IL : interleukine.
IRM : imagerie par résonance magnétique.
LB : lymphocyte B.
LT : lymphocyte T.
MTX : méthotrexate.
PN : polynucléaire neutrophile.
PR : polyarthrite rhumatoïde.
SF-36 : Short Form Health Survey 36.
TNF α : Tumor Necrosis Factor alpha. VS : vitesse de sédimentation.
Références bibliographiques
1. Planète Roche Rhumatologie : L’IL-6, un acteur majeur dans la pathogénie de la polyarthrite rhumatoïde.
2. Oldfield V, Dhillon S, Plosker GL. Tocilizumab: a review of its use in the management of rheumatoid arthritis. Drugs 2009 ; 69 (5) : 609-632.
3. Nishimoto N, Hashimoto J, Miyasaka N, Yamamoto K, Kawai S, Takeuchi T et al. Study of active controlled monotherapy used for rheumatoid arthritis, an IL-6 inhibitor (SAMURAI): evidence of clinical and radiographic benefit from an x ray reader-blinded randomised controlled trial of tocilizumab. Ann Rheum Dis 2007 ; 66 (9) : 1162-7.
4. Nishimoto N, Miyasaka N, Yamamoto K, Kawai S, Takeuchi T, Azuma J et al. Study of active controlled tocilizumab monotherapy for rheumatoid arthritis patients with an inadequate response to methotrexate (SATORI): significant reduction in disease activity and serum vascular endothelial growth

-57-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
factor by IL-6 receptor inhibition therapy. Mod Rheumatolol 2009 ; 19 (1) : 12-19.
5. Maini RN, Taylor PC, Szechinski J, Pavelka K, Bröll J, Balint G et al. Double-blind randomized controlled clinical of the interleukin-6 receptor antagonist, tocilizumab, in European patients with rheumatoid arthritis who had an incomplete response to méthotrexate. Arthritis Rheum 2006 ; 54 (9) : 2817-29.
6. Emery P, Keystone E, Tony HP, Cantagrel A, Van Vollenhoven R, Sanchez A et al. IL-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor biologicals: results from a 24-week multicenter randomised placebo-controlled trial. Ann Rheum Dis 2008 ; 67 (11) : 1516-23.
7. Smolen JS, Beaulieu A, Rubbert-Roth A, Ramos-Remus C, Rovensky J, Alecock E et al. Effect of interleukin-6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): a double-blind, placebo-controlled, randomised trial. Lancet 2008 ; 371 (22) : 987-97.
8. Genovese MC, McKay JD, Nasonov EL, Mysler EF, Da Silva NA, Alecock E, et al. Interleukin-6 receptor inhibition with tocilizumab reduces disease activity in rheumatoid arthritis with inadequate response to disease-modifying antirheu-matic drugs: the tocilizumab in combination with traditional disease-modifying antirheumatic drug therapy study. Arthritis Rheum 2008 ; 58 (10) : 2968-80.
9. Jones G, Sebba A, Gu J, Lowenstein MB, Calvo A, Gomez-Reino JJ et al. Comparison of tocilizumab monotherapy versus methotrexate monotherapy in patients with moderate to severe rheumatoid arthritis: the AMBITION study. Ann Rheum Dis 2010 ; 69 (1) : 88-96.
10. Fleischmann R. Etude LITHE. Communication orale au Congrès de l’ACR 2009 à Philadelphie puis Abstract au Congrès de l’EULAR 2010 à Rome.
11. Nishimoto N, Miyasaka N, Yamamoto K, Kawai S, Takeuchi T, Azuma J. Long-term safety and efficacy of tocilizumab, an anti-interleukin-6 receptor monoclonal antibody, in monotherapy, in patients with rheumatoid arthritis (the STREAM study): evidence of safety and efficacy in a 5-year extension study. Ann Rheum Dis 2008 ; 68 (10) : 1580-4.
12. Bellet D, Dangles-Marie V. Anticorps humanisés en thérapeutique. Médecine Sciences 2005 ; 21 : 1054-62.
13. Weir N, Athural D, Brown D, Foulkes R, Kollias G, Nesbitt A et al. A new generation of high-affinity humanized PEGylated Fab’ fragment anti-tumor necrosis factor alpha monoclonal
antibodies. Therapy 2006 ; 3 (4) : 535-45.
14. Nesbitt A, Fossati G, Bergin M, Stephens P, Stephens S, Foulkes R et al. Mechanism of action of certolizumab pegol (CDP870): in vitro comparison with other anti-tumor necrosis factor α agents. Inflamm Bowel Dis 2007 ; 13 (11) : 1323-32.
15. Chapman AP. PEGylated antibodies and antibody fragments for improved therapy: a review. Advanced Drug Delivery Reviews 2002 ; 54 (4) : 531-5.
16. Duggan ST, Keam SJ. Certolizumab pegol in RA. BioDrugs 2009 ; 23 (6) : 407-17.
17. Taylor PC. Pharmacology of TNF blockade in rheumatoid arthritis and other chronic inflammatory diseases. Current Opinion in Pharmacology 2010 ; 10 (3) : 308-15.
18. Maese PJ. Certolizumab pegol for rheumatoid arthritis: effective in combination with methotrexate or as monotherapy. Int J Clin Rheumatol 2009; 4 (3) : 253-66.
19. Keystone E, van der Heijde D, Mason D, Landewé R, Vollenhoven RV, Combe B et al. Certolizumab pegol plus methotrexate is significantly more effective than placebo plus methotrexate in active rheumatoid arthritis: findings of a fifty-two-week, phase III, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Arthritis Rheum 2008 ; 58 (11): 3319-29.
20. Smolen JS, Landewé EB, Mease P. Efficacy and safety of certolizumab pegol plus methotrexate in active rheumatoid arthritis: the RAPID 2 study. A randomised controlled trial. Ann Rheum Dis 2009 ; 68 (6) : 797-804.
21. Fleischmann R, Vencovsky J, van Vollenhoven RF, Borentein D, Box J, Coteur G et al. Efficacy and safety of certolizumab pegol monotherapy every 4 weeks in patients with rheumatoid arthritis failing previous disease-modifying antirheumatic therapy: the FAST4WARD Study. Ann Rheum Dis 2009 ; 68 (6) : 805-11.
22. Nesbitt A, Lamour S, Mariette X. Le polyéthylène glycol du certolizumab pegol inhibe la dégranulation des mastocytes- Communication affichée.
23. McCluggage L, Scholtz JM. Golimumab: a tumor necrosis factor alpha inhibitor for the treatment of rheumatoid arthritis. Ann Pharmacother 2010 ; 44 (1) : 135-44.
24. Olfield V, Plosker G. Golimumab: in the treatment of rheumatoid arthritis, psoriatic arthritis and ankylosing spondylitis. Biodrugs 2009 ; 23 (2) : 125-35.
25. Smolen JS, Kay J, Doyle M, Landewé R, Matteson EL, Wollenhaupt J, Gaylis N et al. Golimumab in patients with active
rheumatoid arthritis after treatment with tumour necrosis factor alpha inhibitors (Go-After study): a multicentre, randomised, double-blind, placebo-controlled, phase III trial. Lancet 2009 ; 374 (9685) : 210-21.
26. Emery P, Fleischmann RM, Moreland LW, Hsia EC, Strusberg I, Durez P et al. Golimumab, a human anti-tumor necrosis factor α monoclonal antibody, injected subcutaneously every four weeks in methotrexate-naive patients with active rheumatoid arthritis : twenty-four-week results of a phase III, multicenter, randomized, double-blind, placebo-controlled study of golimumab before methotrexate as first-line therapy for early-onset rheumatoid arthritis. Arthritis Rheum 2009 ; 60 (8) : 2272-83.
27. Keystone EC, Genovese MC, Klareskog L, Hsia EC, Hall ST, Miranda PC, Pazdur J et al. Golimumab, a human antibody to tumour necrosis factor alpha given by monthly subcutaneous injections, in active rheumatoid arthritis despite methotrexate therapy: the GO-FORWARD Study. Ann Rheum Dis 2009 ; 68 (6): 789-96.
28. Kremer J, Ritchlin C, Mendelsohn A, Baker D, Kim L, Xu Z et al. Golimumab, a new human anti-tumor necrosis factor α antibody, administered intravenously in patients with active rheumatoid arthritis : Forty-eight-week efficacy and safety results of a phase III randomized, double-blind, placebo-controlled study. Arthritis Rheum 2010 ; 62 (4) : 917-28.
29. Furst DE, Keystone EC, Fleischmann R, Mease P, Breedveld FC, Smolen JS, et al. Updated consensus statement on biological agents for the treatment of rheumatic diseases, 2009. Ann Rheum Dis 2010 ; 69 (suppl I) : i2-i29.
30. Nam JL, Winthrop KL, van Vollhoven RF, Pavelka K, Valesini G, Hensor EM et al. Current evidence for the management of rheumatoid arthritis with biological disease-modifying antirheumatic drugs: a systematic literature review informing the EULAR recommendations for the management of RA. Ann Rheum Dis 2010 ; 69 (6) : 976-86.
31. HAS. Polyarthrite rhumatoïde : diagnostic et prise en charge initiale, septembre 2007. http//www.has-sante.fr ; dernière consultation le 02/10/2010.
32. HAS. Polyarthrite rhumatoïde évolutive grave, avril 2008 http//www.has-sante.fr ; dernière consultation le 02/10/2010.
33. HAS. Polyarthrite rhumatoïde : Prise en charge en phase d’état, septembre 2007. http//www.has-sante.fr ; dernière consultation le 02/10/2010.
34. Fautrel B, Constantin A, Morel J, Vittecoq O, Cantagrel A, Combe B. Recommandations de la Société Française de Rhumatologie pour l’utilisation des agents anti-TNFα chez les

-58-
Dossier du CNHIM, 2010, XXXI, 5
Polyarthrite rhumatoïde (2ème partie) : nouvelles biothérapies, tocilizumab, certolizumab, golimumab
personnes souffrant de polyarthrite rhumatoïde. Revue du Rhumatisme 2006, 73 : 726-735.
35. Sellam et al. Predictive factors of response to rituximab in rheumatoid arthritis with inadequate response or intolerance to anti-TNF: Data from the smart trial. Eular 2010.
36. Gottenberg JE, Ravaud P et al. Prospective follow up of rituximab treatment in 2000 patients with refractory rheumatoid arthritis included in the air registry: risk factors of severe infections. Eular 2010-08-22.
37. Kukar M, Petryna O, Efthimiou P. Biological targets in the treatment of rheumatoid arthritis: a comprehensive review of current and in-development biological disease modifying anti-rheumatic drugs. Biologics 2009 ; 3 : 443-57.
38. Genovese MC, Van den Bosch F, Roberson SA, Bojin S, Ryan P. LY2439821, a humanized anti-interleukin-17 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: a phase I randomized, double-blind, placebo controlled, proof-of-concept study. Arthritis Rheum 2010 ; 62 (4) : 929-39.
39. Churchman SM, Ponchel F. Interleukin-7 in Rheumatoid arthritis. Rheumatology 2008; 47 : 753-9.
40. Sharp JT, Tsuji W, Ory P, Harper-Barek C, Wang H, Newmark R. Denosumab prevents metacarpal shaft cortical bone loss in patients with erosive rheumatoid arthritis. Arthritis Care Res 2010; 62 : 537-44.
41. Ostergaard M, Baslund B, Rigby W, Rojkovich B, Jorgensen C, Dawes PT et al. Ofatumumab, a human anti-CD20
monoclonal antibody, for treatment of rheumatoid arthritis with an inadequate response to one or more disease-modifying antirheumatic drugs. Arthritis Rheum 2010 ; 62 : 2227-38.
42. Documents des laboratoires : Tocilizumab (ROACTEMRA® – laboratoire
Chugaï Roche) ; certolizumab (CIMZIA® – laboratoire UCB) ; golimumab (SIMPONI® – laboratoire Schering Plough).
43. Fiches du CRI : Anti-TNF ; Tocilizumab.
44. Fiches de Bon Usage des médicaments HAS : tocilizumab (ROACTEMRA®) ; certolizumab (CIMZIA®).
Annexes
Annexe 1. Critères d’évaluation de la réponse thérapeutique des malades traités (d’après 33)
Amélioration du DAS 28 Par rapport à la mesure de base Nouvelle valeur du DAS 28 atteinte durant le suivi > 1,2 >0,6 et < ou = 1,2 < ou = 0,6
2,6<DAS 28≤3,2
PR d’activité faible Bonne réponse Réponse modérée Absence de réponse
3,2<DAS 28<5,1
PR d’activité modérée Réponse modérée Réponse modérée Absence de réponse
DAS>5,1
PR d’activité forte Réponse modérée Absence de réponse Absence de réponse
Annexe 2. Interprétation clinique de l’indice fonctionnel HAQ-DI (31)
Variation par rapport à la valeur initiale du score HAQ-DI
HAQ -DI Catégorie 1 Catégorie 2 Catégorie 3
Amélioration < ou = - 0,25 < ou = - 0,3 < ou = - 0,5
Pas de changement > - 0,25 à < 0,25 > - 0,3 à < 0,3 > - 0,5 à < 0,25
Pire > ou = 0,25 > ou = 0,3 > ou = 0,5

-59-
Dossier du CNHIM, 2010, XXXI, 5
Fiche IAM : inhibiteurs de la phosphodiestérase de type 5
Niveaux de contrainte
*cf. au verso : conduite à tenir spécifique.
Association à prendre en compte (APEC) Association Déconseillée (ASDEC)
Interactions médicamenteuses des inhibiteurs de la phosphodiestérase de type 5 : sildénafil, tadalafil et vardénafil
Amélie Hugon, Isabelle Federspiel, Pierrick Bedouch, Benoît Allenet, Jean Calop
Pôle pharmacie, CHU de Grenoble
Remerciements : I. Fusier (Paris), MC. Husson (Paris), C. Tollier (Paris).
Précaution d’emploi (PE)
Hypotension orthostatique
� = IAM de la classe � = IAM spécifique de la substance en sus des IAM de la classe
Interactions médicamenteuses des inhibiteurs de la phosphodiestérase de type 5 ����
D’après : Thesaurus Afssaps, Juin 2009
Alpha bloquants à visée urologique (sauf doxazosine)*
Antihypertenseurs alpha-bloquants* Clarythromycine* Erythromycine*
Doxazosine Ritonavir* (sauf avec le vardénafil)
Dérivés nitrés
Association Contre-indiquée (CI)
Itraconazole* Kétoconazole*
sildénafil
et ta
dalafil
Josamycine*
Interactions médicamenteuses du sildénafil ����
Hypotension orthostatique
Itraconazole Kétoconazole
chez l'homme de plus de 75 ans
Itraconazole Kétoconazole
chez l'homme jusqu'à 75 ans
vardénafil
Inhibiteurs de protéase
Inhibiteurs de la protéase (sauf ritonavir)*

-60-
Dossier du CNHIM, 2010, XXXI, 5
Fiche IAM : inhibiteurs de la phosphodiestérase de type 5
1. Commentaires généraux Les inhibiteurs de la phosphodiestérase de type 5 sont prescrits dans deux indications distinctes ; les modalités de prise et de dispensation différant selon les indications : - Traitement du dysfonctionnement érectile : CIALIS® (tadalafil) 2.5, 5, 10 et 20 mg, VIAGRA® (sildénafil) : 25, 50 et 100mg, LEVITRA® (Vardénafil) : 5, 10 et 20 mg, à prendre avant toute activité sexuelle prévue (médicaments inscrits sur liste I et non remboursés)
- Prise en charge de l’hypertension artérielle pulmonaire chez les malades en classe fonctionnelle II et III (selon la classification de l'OMS). Le REVATIO®
(sildénafil) est utilisé à la dose de 20 mg 3 fois par jour avec un délai de 6 à 8h entre chaque prise. Le traitement doit être uniquement initié et contrôlé par un médecin expérimenté dans la prise en charge de l'hypertension artérielle pulmonaire. (Ce médicament est soumis à prescription hospitalière et est inscrit sur la liste de rétrocession).
2. Mécanismes des IAM
2.1. IAM dépendantes d’une interaction avec le cytochrome P450
- Inhibition enzymatique du métabolisme hépatique des inhibiteurs de la phosphodiestérase de type 5 par : itraconazole, kétoconazole, érythromycine, clarithromycine, josamycine, inhibiteurs de la protéase (amprénavir, atazanavir, darunavir, fosamprénavir, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir, tipranavir), d’où augmentation des concentrations sanguines en inhibiteurs de la phosphodiestérase de type 5, majorant les effets indésirables type hypotension orthostatique, céphalées, rougeurs, dyspepsie, troubles de la vision, sensations vertigineuses.
2.2. IAM indépendantes d’une interaction avec le cytochrome P450 - Addition des effets hypotenseurs observés avec les alpha bloquants à visée urologique (alfuzosine, doxazosine, prazosine, tamsulozine, térazosine) et antihypertenseurs alpha bloquants (prazosine, trimazosine, urapidil) notamment chez le sujet âgé.
- Addition des effets hypotenseurs et risque d’aggravation de l'état d'ischémie myocardique (effets synergiques) avec les dérivés nitrés et apparentés (dinitrate d'isosorbide, isosorbide, linsidomine, molsidomine, nicorandil, trinitrine) sous quelque forme que se soit.
2.3. IAM(s) à suivre, non mentionnée(s) dans le thesaurus AFSSAPS - Induction enzymatique du métabolisme hépatique des inhibiteurs de la phosphodiesterase de type 5 par le bosentan, d’où diminution des concentrations sanguines d’inhibiteurs de la phosphodiesterase de type 5. * RCP Rectificatif AMM européenne 07/07/2009
* Mutual pharmacokinetic interactions between steady-state bosentan and sildenafil. Eur J Clin Pharmacol 2008 Jan ; 64 (1) : 43-50.
2.4. Cytochromes P450 et inhibiteurs de la phosphodiesterase de type 5 Cf. tableaux page 61.
3. Conduites à tenir
3.1. Générale Surveillance clinique et adaptation éventuelle de la posologie du médicament concerné.
3.2. Spécifiques (substances actives avec *)
- Alpha-bloquants à visée urologique, antihypertenseurs alpha-bloquants, inhibiteurs de la protéase (sauf ritonavir), clarithromycine, érythromycine, itraconazole, kétoconazole : Débuter le traitement aux posologies minimales recommandées et adapter progressivement les doses si besoin. - Ritonavir : Ne pas dépasser la posologie minimale de l'inhibiteur de la phosphodiestérase de type 5 toutes les 48 heures. - Clarithromycine, érythromycine : en cas d’association avec le vardénafil, diminuer la posologie de l’inhibiteur de la phosphodiestérase de type 5.
Interactions médicamenteuses des inhibiteurs de la phosphodiestérase de type 5 : sildénafil, tadalafil et vardénafil
Commentaires, mécanismes, conduites à tenir

-61-
Dossier du CNHIM, 2010, XXXI, 5
Fiche IAM : inhibiteurs de la phosphodiestérase de type 5
Tableau 1 : Substrats des cytochromes P450 (d’après http//www.pharmacoclin.ch)
Substance active 1A2 2B6 2C9 2C19 2D6 2E1 3A4
Tadalafil Sildénafil Vardénafil Amprénavir Bosentan Clarithromycine Erythromycine Indinavir Itraconazole Kétoconazole Lopinavir Nelfinavir Ritonavir Saquinavir Tamsulozine Voie Métabolique : Majeure Mineure
Tableau 3: Inducteurs des cytochromes P450
(d’après http//www.pharmacoclin.ch)
Substance active 1A2 2B6 2C9 2C19 2D6 2E1 3A4
Amprénavir Bosentan Ritonavir
Inducteur : Puissant Modéré
Tableau 2 : Inhibiteurs des cytochromes P450
(d’après http//www.pharmacoclin.ch)
Substance active 1A2 2B6 2C9 2C19 2D6 2E1 3A4
Amprénavir Clarithromycine Erythromycine Indinavir Itraconazole Kétoconazole Lopinavir Nelfinavir Ritonavir Saquinavir
Inhibiteur : Puissant Modéré
Pour une bonne utilisation des fiches Interactions médicamenteuses : Husson MC. Interactions médicamenteuses : modalités d’utilisation des fiches. Doss CNHIM 2006 ; XXVII (2) : 37-40.

-62-
Dossier du CNHIM, 2010, XXXI, 5
Fiche IAM : inhibiteurs de la phosphodiestérase de type 5
Cas clinique n°1
Cas clinique n°2
Malade et contexte
Mr S.T, 85 ans
Antécédents :
Adénome de la prostate traité par XATRAL LP 10 MG®
(alfuzosine) 1 comprimé le soir. Histoire de la maladie : Dysfonctionnement érectile. Ordonnance : VIAGRA® (sildénafil) 100 mg : une fois dans la journée selon les besoins du malade.
Problématique médicamenteuse
Evénement intercurrent : Angine bactérienne traitée par macrolides : ZECLAR®
Interaction médicamenteuse : La clarythromycine est un inhibiteur enzymatique qui peut en cas d’association avec un inhibiteur des phosphodiestérase de type 5, majorer le risque d’hypotension orthostatique.
Malade et contexte Mr A.P., 62ans
Antécédents : Trouble érectile ayant motivé une prescription itérative de CIALIS® 10 mg (tadalafil). Histoire de la maladie : Consulte son médecin traitant pour fièvre et douleurs à la déglutition. Mise en évidence d’une angine à streptocoque A dans un contexte d’allergie avérée aux pénicillines. Ordonnance :
ZECLAR® (clarythromycine) 250 mg : 2 comprimés par
jour pendant 10 jours.
Problématique médicamenteuse
Evénement intercurrent : Mise en place d’un traitement par inhibiteur des phosphodiestérase de type 5. Interaction médicamenteuse : L’association d’un alpha bloquant à visée urologique et d’un inhibiteur de la phosphodiestérase peut majorer le risque d’hypotension.
Intervention pharmaceutique
Afin de minimiser l'éventuelle survenue d'une hypotension orthostatique, le pharmacien a recommandé une initiation du traitement par sildénafil à dose plus faible. Il a expliqué au malade les signes cliniques d’une hypotension, pour lui permettre de les reconnaitre, et lui a conseillé d’arrêter le VIAGRA® en cas d’apparition de ces signes et de consulter son médecin traitant.
Intervention pharmaceutique Le pharmacien a averti le médecin quant à l’existence de cette interaction médicamenteuse. Il a conseillé, le temps du traitement antibiotique, d’éviter la prise de CIALIS® afin de prévenir l’apparition des effets indésirables.
Interactions médicamenteuses des inhibiteurs de la phosphodiestérase de type 5
Quatre exemples d’interventions pharmaceutiques

-63-
Dossier du CNHIM, 2010, XXXI, 5
Fiche IAM : inhibiteurs de la phosphodiestérase de type 5
Cas clinique n°3
Malade et contexte Mr T. B, 56 ans
Antécédents : Malade VIH positif traité par COMBIVir® (lamivudine + zidovudine) 1 cpr 2 fois par jour, KALETRA® (lopinavir, ritonavir) 2 cpr 2 fois par jour.
Histoire de la maladie : Problème érectile fréquent. Sur les conseils de son entourage, la femme du malade souhaiterait que son mari essaie le CIALIS® (tadalafil) et décide de se procurer ce traitement via internet. Ordonnance : CIALIS® (tadalafil) 10 mg : une fois dans la journée selon les besoins du malade.
Problématique médicamenteuse
Evénement intercurrent : automédication par inhibiteur de la phosphodiestérase de type 5 sans avis médical. La femme du malade raconte à son pharmacien sa démarche lors du renouvellement du traitement VIH. Interaction médicamenteuse : L’antiprotéase ritonavir est un puissant inhibiteur du cytochrome P450 pouvant entraîner une augmentation importante des concentrations plasmatiques de l'inhibiteur de la phosphodiestérase de type 5, avec risque d'hypotension sévère.
Intervention pharmaceutique
Le dialogue avec la femme du malade a permis au pharmacien de mettre en évidence cette automédication à l’origine d’une interaction médicamenteuse. Il a recommandé au couple de ne pas utiliser ce traitement. Il l’a informé que ce médicament doit être prescrit par un médecin du fait des interactions médicamenteuses potentielles. Le pharmacien leur a recommandé de consulter leur médecin traitant pour définir la conduite à tenir pour la prise en charge des dysfonctions érectiles chez ce malade.
Cas clinique n°4
Malade et contexte
Mr T.F, 63 ans
Antécédents : Hypertension artérielle pulmonaire traitée par REVATIO® (sildénafil) 20 mg : 3 cpr par jour Histoire de la maladie : Augmentation des fréquences de mictions diurnes et nocturnes l’amenant à consulter son médecin traitant. Ordonnance : OMIX LP® (tamsulozine) 4 mg : 1 gélule par jour.
Intervention pharmaceutique Le pharmacien a averti le médecin de la majoration des effets hypotenseurs et propose un traitement de l’HBP par une autre classe médicamenteuse que les alpha-bloquants à visée urologique. Il a recommandé un traitement par les inhibiteurs de la 5-alpha réductase ou par les extraits de plantes.
Problématique médicamenteuse
Evénement intercurrent : hypertrophie bénigne de la prostate (HBP) avec instauration d’un traitement par tamsulozine. Interaction médicamenteuse : L’association d’un alpha bloquant à visée urologique associé à un inhibiteur des phosphodiestérase de type 5 augmente le risque d’hypotension orthostatique.
Interactions médicamenteuses des inhibiteurs de la phosphodiestérase de type 5
Quatre exemples d’interventions pharmaceutiques (suite)

-64-
Dossier du CNHIM, 2010, XXXI, 5
Fiche IAM : inhibiteurs de la phosphodiestérase de type 5

-65-
Dossier du CNHIM, 2010, XXXI, 5
Résumés des derniers numéros parus
N°3, 2010 : Hyperammoniémies par déficit enzymatique du cycle de l’urée : place du benzoate de sodium
Les hyperammoniémies par déficit enzymatique du cycle de l’urée (UCD : Urea Cycle Disorders) sont des maladies héréditaires rares du métabolisme. Six enzymes sont concernées : 3 mitochondriales. Le déficit le plus fréquent et le mieux décrit à ce jour est le déficit en OTC, qui obéit à un mode de transmission lié à l’X. Les 5 autres déficits (NAGS, CPS-I, ASS, ASL, ARGa) sont de transmission autosomique récessive. L’incidence de toutes les maladies dues à ces différents déficits a été estimée à 1 / 8200. Les UCD peuvent se révéler à tout âge. La décompensation aiguë est caractérisée par des troubles digestifs, neurologiques et de la conscience pouvant aller jusqu’au coma. L’hyperammoniémie se définit par une augmentation de la concentration en ion ammonium plasmatique > 100 µmol/L chez le nouveau-né, et > 50 µmol/L chez l’enfant et l’adulte. En France le traitement des hyperammoniémies par UCD consiste à administrer : - dans les formes aiguës en urgence le benzoate de sodium AP-HP, 1g/10 mL, solution à diluer pour perfusion (préparation hospitalière de l’AGEPS). Est aussi utilisé l’AMMONUL® 10% 10% solution injectable (mélange équimolaire de phénylacétate de sodium et de benzoate de sodium), commercialisé aux USA et disponible avec une ATU nominative en France.
- en relais de ce traitement d’urgence, un traitement per os de benzoate de sodium en gélules (préparation magistrale) associé si nécessaire à l’AMMONAPS® comprimé 500 mg et granulé 940 mg/g (phénylbutyrate de sodium) dans le traitement au long cours. Les traitements d’entretien au long cours associent à ces traitements épurateurs des mélanges d’acides aminés essentiels (L-arginine, citrulline, ornithine, L-carnitine). Le CARBAGLU® (acide carglumique) est le traitement spécifique des déficits en NAGS et de certains déficits en CPS. La prise en charge des malades doit être réalisée par une équipe spécialisée pluridisciplinaire. Une enquête d’usage sur le benzoate de sodium AP-HP 1 g/10 mL, solution injectable pour perfusion, a été menée en juin 2009. Elle confirme l’intérêt pour ce médicament malgré la pauvreté d’études cliniques, mais seule une enquête rétrospective pourrait permettre d’évaluer le nombre de malades atteints d’UCD avec hyperammoniémie, et l’impact réel du benzoate de sodium. La mise en œuvre d’études cliniques prospectives comparatives, en France et en Europe, pourrait permettre de déterminer le rapport bénéfice/risque des différents médicaments disponibles, leur efficacité relative et l’intérêt de leur association.
Curares et décurarisation : place du sugammadex Les curares sont utilisés pour faciliter l’intubation trachéale et l’immobilité du malade lors d’interventions chirurgicales. Ils permettent une relaxation des muscles striés en inhibant la transmission neuromusculaire. Lors du monitorage de la curarisation et de la décurarisation du malade, si la décurarisation complète ne peut être affirmée, un anticholinestérasique peut être administré. Après la néostigmine, un nouveau décurarisant le sugammadex - BRIDION® est arrivé fin 2008 sur le marché européen et français. Le chlorure de suxaméthonium est un ammonium quaternaire composé de deux molécules d’acétylcholine, il agit donc comme un agoniste du récepteur à l’acétylcholine. Les curares non dépolarisants sont des ammoniums quaternaires composés d’au moins un atome d’azote chargé positivement leur permettant de se fixer à une sous unité α des récepteurs nicotiniques de l’acétylcholine. Ils agissent comme antagonistes compétitifs de ces récepteurs selon la loi d’action de masse. Le monitorage pendant et après l’administration de curares est fortement recommandé. Le délai de récupération est très variable
selon le curare utilisé, le malade, les autres anesthésiques employés. Il est donc peu prévisible et le monitorage instrumental de la curarisation est indispensable. L’antagonisation de la curarisation est recommandée si la décurarisation complète ne peut être affirmée. Elle a pour but d’accélérer la décurarisation lorsque celle-ci a déjà commencé. Le sugammadex (BRIDION®) a une forte affinité pour les curares stéroïdiens. Son indication est la décurarisation chez l’adulte après un bloc neuromusculaire induit par rocuronium ou vécuronium. La dose dépend du degré du bloc neuromusculaire à décurariser ; elle est indépendante du protocole anesthésique. Le sugammadex est administré en bolus IV unique. La néostigmine a une efficacité limitée et présente des effets indésirables. L’antagoniste idéal des curares non dépolarisants serait capable de reverser rapidement un bloc neuromusculaire, indépendamment de sa profondeur au moment de la décurarisation et de la technique d’anesthésie employée. Le sugammadex est une réelle innovation pharmacologique, son principal inconvénient est de n’agir que sur le rocuronium et le vécuronium.
N°4, 2010 : Polyarthrite rhumatoïde (1ère partie) : nouvelles biothérapies ciblant les cellules du système immunitaire, rituximab et abatacept
La polyarthrite rhumatoïde (PR) est un rhumatisme inflammatoire chronique touchant plusieurs articulations et associé à une inflammation de la membrane synoviale. C’est une pathologie auto-immune évoluant par poussées et selon des schémas de gravité hétérogènes. En l’absence de traitement, la PR entraîne des destructions articulaires et des déformations. L’origine de cette pathologie est multifactorielle. Le diagnostic, avant tout clinique, doit être le plus précoce possible. Une fois installée, la PR s’aggrave progressivement avec une extension des atteintes articulaires puis des atteintes extra-articulaires de plus en plus fréquentes pouvant mettre en jeu le pronostic vital du malade. En 1987, l’American College of Rheumatology (ACR) a proposé 7 critères de classification de la PR permettant la définition d’un profil de malades. En 2009 de nouveaux critères - les ACR 2009 - sont des critères de diagnostic précoce de la maladie. La réponse thérapeutique sur l’activité de la maladie peut être jugée par différents critères permettant un suivi du traitement de fond de la PR : l’ACR 20, l’amélioration du DAS 28, la mesure de la qualité de vie fonctionnelle (HAQ-DI, SF-36), l’évaluation de la fatigue ressentie par le malade (FACIT), des critères radiologiques. La prise en charge thérapeutique de la maladie, la plus précoce possible, consiste en : - traitements symptomatiques : anti-inflammatoires non stéroïdiens ou corticoïdes, antalgiques de niveau I et II, injection locale de dérivés cortisoniques ; - traitements de fond, seuls ou en association : méthotrexate (médicament de référence), sulfasalazine, léflunomide, hydroxychlo-roquine, azathioprine, D-pénicillamine, ciclosporine et sels d’or ; - depuis une dizaine d’années, les biothérapies dont les premières cibles thérapeutiques ont visé les cytokines (TNF-α et IL-1).
L’utilisation clinique des anti-TNFα (adalimumab, étanercept et infliximab) s’est progressivement affinée. L’anakinra, antagoniste du récepteur de l’IL-1, a montré une efficacité satisfaisante mais avec une administration quotidienne par voie sous-cutanée, plus contraignante. Les nouvelles biothérapies visent des cellules immunitaires, les lymphocytes B avec le rituximab, les lymphocytes T avec l’abatacept ; ces deux médicaments sont traités dans cet article. Le rituximab est un anticorps monoclonal dirigé contre le CD20, présent sur tous les lymphocytes B matures. En 2006, le rituximab a obtenu l’AMM dans la prise en charge des PR actives et sévères en échec d’au moins un anti-TNF, selon le schéma d’administration de 2 perfusions de 1 g à 14 jours d’intervalle. Des données récentes préconisent un intervalle de 6 mois entre 2 cures. L’abatacept est un immunosuppresseur, modulateur sélectif de la costimulation des lymphocytes T, en mimant l’action physiologique du CTLA-4. Il est indiqué, en association au méthotrexate, dans le traitement de la PR active, modérée à sévère, en cas de réponse inadéquate ou d’intolérance au traitement de fond, dont au moins un anti-TNF. Il en résulte une réduction de la progression des dommages structuraux et une amélioration des capacités fonctionnelles. Les thérapeutiques médicamenteuses utilisées dans la PR se sont étoffées plus récemment avec de nouvelles biothérapies visant des cytokines : l’IL-6 avec le tocilizumab, le TNF-α avec le certolizumab et le golimumab. Ces trois nouveaux médicaments seront évalués dans le prochain Dossier du CNHIM N°5-2010, avec les stratégies thérapeutiques actuelles.
Résumés des derniers numéros parus Dossier du CNHIM

-66-
Dossier du CNHIM, 2010, XXXI, 4
Sommaire
Année 2001-Tome XXII N°1-2 Médicaments utilisés en cancérologie (4ème éd.) N°3 Traitements de la bronchiolite à VRS : place du palivizumab Perchlorate de potassium et pathologies thyroïdiennes N°4 Traitement du diabète de type 2 : place des nouveaux
antidiabétiques oraux N°5 Traitements d’éradication de Helicobacter pylori N°6 Traitement de la maladie de Crohn
Année 2002-Tome XXIII N°1 Olanzapine dans le traitement de la schizophrénie Hémine humaine dans le traitement des crises aiguës de
porphyries hépatiques N°2 Prévention des accidents ischémiques cérébraux par les
antiagrégants plaquettaires et les anticoagulants N°3 Tocolytiques et menace d’accouchement prématuré N°4 Milnacipran, venlafaxine, mirtazapine dans le traitement des
épisodes dépressifs majeurs N°5-6 Principales associations d’antirétroviraux dans le traitement
des infections à VIH
Année 2003-Tome XXIV N°1 Algasidases (alfa, bêta) dans le traitement de la maladie de
Fabry Valganciclovir Fludrocortisone N°2 Coxibs : inhibiteurs sélectifs de la COX-2 N°3-4 Facteurs antihémophiliques : traitement substitutif de
l’hémophilie A et B N°5 Polyarthrite rhumatoïde : stratégie thérapeutique N°6 Escarre, ulcère, pied diabétique : pansements et
biomatériaux. Aide à la cicatrisation
Année 2004-Tome XXV N°1 Prévention du rejet aigu de greffes rénales place des
anticorps monoclonaux N°2 Ostéoporose : place des bisphosphonates et du SERM N°3 Sepsis sévère et choc septique : données actuelles - Place de
la protéine C N°4-5 Anticancéreux : utilisation pratique, 5ème édition N°6 Candidoses et aspergilloses invasives : stratégie
thérapeutique
Année 2005-Tome XXVI N°1 Statines et prévention des risques cardiovasculaires N°2 Hypertension artérielle pulmonaire : stratégies de prise en
charge Ibuprofène injectable dans le traitement de la persistance du
canal artériel N°3 Maladie d’Alzheimer : traitements médicamenteux N°4-5 Médicaments radiopharmaceutiques : 2ème édition
Année 2006-Tome XXVII N°1 Trouble déficitaire de l’attention avec hyperactivité : prise en
charge thérapeutique N°2 Syndrome de lyse tumorale : prise en charge Mucopolysaccharidose de type I : traitement actuel Interactions : anticoagulants oraux, antalgiques morphiniques N°3 Insulines : utilisation pratique Interactions : macrolides sauf spiramycine N°4 Cystinose et cystéamine Interactions : apparentés macrolides et spiramycine ;
antirétroviraux ; immunosuppresseurs N°5-6 Maladie thromboembolique : stratégies thérapeutiques
préventives et curatives Interactions : anticancéreux ; hypolipémiants ; cyclines ;
fluoroquinolones ; sulfamides antibactériens
Année 2007-Tome XXVIII
N°1 Produits de contraste pour imagerie par résonance magnétique
Interactions: bêtalactamines, aminosides, antituberculeux N°2 Maladie de Gaucher : traitements actuels
Interactions : antagonistes des récepteurs de l’angiotensine II, bêtabloquants, inhibiteurs de l’enzyme de conversion
N°3 Mucoviscidose : place de l’antibiothérapie inhalée Interactions : antidépresseurs imipraminiques, inhibiteurs
sélectifs de la recapture de la sérotonine ; antifongiques azolés.
N°4-5 Thérapie photodynamique Eptacog alpha, rFVIIa, NOVOSEVEN® : 1ère partie Interactions : anticonvulsivants, antiparkinsoniens, anti
Alzheimer N°6 Migraine : stratégies thérapeutiques Eptacog alpha, rFVIIa, NOVOSEVEN® : 2ème partie :
Traumatologie
Année 2008-Tome XXIX N°1 Immunoglobulines humaines normales sous-cutanées :
traitement substitutif Fluoroquinolones : place de la lévofloxacine et de la
moxifloxacine Interactions : Antisécrétoires antihistaminiques H2 et inhibiteurs de la pompe à protons
N°2 Solutions de conservation d’organes Eptacog alpha, NOVOSEVEN® : 3ème partie : Gynéco-obstétrique
N°3 Adhérences : prévention en chirurgie digestive et gynécologiques
Eptacog alpha, rFVIIa, NOVOSEVEN® : 4ème partie : Chirurgie cardiaque
N°4 Adhésion tissulaire, hémostase locale et consolidation : traitements locaux
Interactions : cisapride et diphémanil N°5-6 Anticancéreux : utilisation pratique, 6ème édition
Année 2009-Tome XXX N°1 Prescrire en dénomination commune pour sécuriser les
prescriptions : intérêt du médicament virtuel Eptacog alpha, rFVIIa, NOVOSEVEN® : 5ème partie :
Hématologie Interactions : Insuline et antidiabétiques oraux ;
Antimigraineux : triptans, alcaloïdes de l’ergot de seigle N°2 Mucopolysaccharidoses de type II et de type VI :
traitements enzymatiques substitutifs Eptacog alpha, rFVIIa, NOVOSEVEN® : 6ème partie :
Utilisation en hépato-gastro-entérologie Interactions : antihypertenseurs alpha-bloquants ; alpha-
bloquants à visée urologique N°3 Hormones de croissance
Interactions : inhibiteurs des tyrosines kinases ; topiques gastro-intestinaux, antiacides et charbon
N°4 Chélateurs du fer Interactions médicamenteuses cliniquement significatives : principaux mécanismes
N°5 Erythropoïétines ; 1ère partie : utilisations AMM Eculizumab et hémoglobinurie paroxystique nocturne Interactions : glucocorticoïdes, minéralocorticoïdes N°6 Erythropoïétines ; 2ème partie : utilisations hors AMM Interactions : héparines
Année 2010-Tome XXXI N°1 La vaccination antigrippale 2009-2010 Nutrition parentérale pédiatrique : une nouvelle
gamme de solutions Interactions : acide acétylsalicylique et paracétamol ;
anti-inflammatoires non stéroïdiens N°2 Purpura thrombopénique auto-immun : place des agonistes du récepteur de la thrombopoïétine Interactions : Estrogènes et progestatifs N°3 Hyperammoniémie par déficit enzymatique du cycle de
l’urée : place du benzoate de sodium dans la stratégie thérapeutique
Curares et décurarisation : place du sugammadex N°4 Polyarthrite rhumatoïde (1ère partie) : nouvelles
biothérapies ciblant les cellules du système immunitaire, rituximab et abatacept
Au sommaire de Dossier du CNHIM depuis 2001

Je suis : � Pharmacien � Médecin � Autre : ___________________________
ABONNEMENT 2010
� Je souhaite m'abonner pour un an à la revue Dossier du CNHIM 6 numéros par an
Etablissements de soins, facultés et particuliers : France (tva 2.10 %) � � HT : 195.89 € - TTC : 200 € Etranger � � 220 €
Laboratoires pharmaceutiques, organismes : France (tva 2.10 %) � � HT : 391.78 € - TTC : 400 € Etranger � � 420 €
VENTE AU NUMERO 2010*
� Je souhaite acquérir un (des) exemplaire(s) de la revue Dossier du CNHIM (le prix du numéro spécial est multiplié par deux)
Etablissements de soins, facultés et particuliers : France (tva 2.10 %) � � HT : 39.18 € - TTC : 40 € Etranger � � 60 €
Laboratoires pharmaceutiques, organismes : France (tva 2.10 %) � � HT : 78.36 € - TTC : 80 € Etranger � � 100 €
* Pour les numéros parus avant 2006, veuillez nous contacter. Tarifs dégressifs selon nombre d’exemplaires, veuillez nous contacter.
Veuillez noter..........exemplaire(s) du N°.........................soit...............€ Veuillez noter..........exemplaire(s) du N°.........................soit...............€
Adresse de facturation : Nom : Adresse : Tél : Fax : A retourner accompagné d’un chèque du montant correspondant à l’ordre du CNHIM. Adresse de livraison si différente (envoi par courrier courant selon La Poste) : Nom : Adresse :
CNHIM - Hôpital de Bicêtre – 78, rue du Général Leclerc BP 11 - 94272 Le Kremlin Bicêtre cedex – Association loi 1901 Tél. : +33 (0)1 46 58 07 16 - Fax : +33 (0)1 46 72 94 56 - Courriel : [email protected]
N° SIRET 318 762 051 00023 – APE 913 E – C.C. BANQUE POPULAIRE DU NORD 00-47302-190-4