
Conservatisme phylogénétique des traits fonctionnels … · Chalmandrier Loïc Rapport de stage...
31
Chalmandrier Loïc Rapport de stage Conservatisme phylogénétique des traits fonctionnels végétaux Un test utilisant un échantillon de la Flore Alpine Encadrant : Sébastien Lavergne Laboratoire d'acceuil : Laboratoire d'Ecologie Alpine (UMR 5553) 1
Transcript of Conservatisme phylogénétique des traits fonctionnels … · Chalmandrier Loïc Rapport de stage...

Chalmandrier Loïc
Rapport de stage
Conservatisme phylogénétique des traits fonctionnels végétaux
Un test utilisant un échantillon de la Flore Alpine
Encadrant : Sébastien LavergneLaboratoire d'acceuil : Laboratoire d'Ecologie Alpine (UMR 5553)
1

Table des matièresIntroduction..........................................................................................................................................3Matériel et Méthode.............................................................................................................................6
I Acquisition des données sur les traits fonctionnels.......................................................................6II Analyse phylogénétique...............................................................................................................6
1. Arbre........................................................................................................................................62. Outils statistiques....................................................................................................................6
1. Le et (Pagel 1999, Verdù 2006) : ...........................................................................62. Orthogramme (Ollier et al. 2005).......................................................................................73. Divergence et contribution des noeuds (Moles et al. 2005) : ............................................84. Phylogenetic least squares model (Martins et Hansen 1997 ; RossIbarra 2007) ou PGLS................................................................................................................................................9
Résultats..............................................................................................................................................10I et de Pagel.......................................................................................................................10II Orthogramme.............................................................................................................................11
1. SLA........................................................................................................................................112. LDMC...................................................................................................................................12
III Amplitude de divergence et contribution..................................................................................13IV Etude de la corrélation entre caractères....................................................................................16
Discussion...........................................................................................................................................17Patron évolutif du SLA et du LDMC.............................................................................................17Corrélation des deux patrons d'évolution.......................................................................................17Critique de la démarche de Moles..................................................................................................17Comparaison des trois méthodes....................................................................................................18
Conclusion..........................................................................................................................................19Bibliographie......................................................................................................................................20Glossaire.............................................................................................................................................21Annexes..............................................................................................................................................22
Annexe 1 : Démarche mathématique de Moles.............................................................................221. Estimation des états ancestraux (Felsenstein 1985)...............................................................222. Calcul de l'amplitude de divergences....................................................................................223.3 Calcul de la contribution....................................................................................................23
Annexe 2 : Noeuds mis en avant par la méthode d'Ollier et de Moles..........................................24Annexe 3 : Scripts..........................................................................................................................26
Démarche de Pagel....................................................................................................................26Démarche de Moles...................................................................................................................28
2

IntroductionFace aux enjeux majeurs que représentent les changements environnementaux actuels (réchauffement climatique, modifications de l'usage des terres), il est important de décrire les êtres vivants en termes de traits fonctionnels afin de prédire les changements dans la distribution de la biodiversité et le fonctionnement des écosystèmes. En effet, les traits fonctionnels, en particulier chez les végétaux, permettent à la fois d’étudier la réponse des espèces aux gradients environnementaux et leurs changements , et de prédire l’effet de ces mêmes espèces sur le fonctionnement des écosystèmes (par exemple la production de biomasse ou la décomposition de la litière dans les communautés végétales...) Plus précisément, on peut définir un trait fonctionnel comme étant un trait morphologique, physiologique ou phénologique ayant un impact indirect sur la fitness de la plante via ses effets sur la croissance, la reproduction et la survie des individus (Violle et al. 2007). Il a été démontré (Díaz et al. 2004) que certains traits fonctionnels dits « soft », c'estàdire facilement mesurables et donc plus susceptibles de constituer de larges jeux de données, peuvent ainsi se corréler à des traits dits « hard » tel le taux de croissance ou le taux de azote par feuille, directement liés à la fitness des individus. Au sein des écosystèmes, la diversité fonctionnelle, c'estàdire la largeur du spectre des valeurs de traits de l'ensemble des individus de l'écosystème, constitue un élément essentiel de leur bon fonctionnement et de leur stabilité (Díaz et Cabido 2001). S’il est maintenant à peu près établi que la diversité fonctionnelle joue un rôle important dans le fonctionnement des écosystèmes, les mécanismes évolutifs qui sont à l’origine de la diversité fonctionnelle chez les végétaux sont encore mal connus. En particulier, l’utilisation de phylogénies, c'estàdire d'arbres traduisant des relations de parenté entre espèces ayant donc un ancêtre commun hypothétique, permet de tester si les traits fonctionnels ont connu de fortes variations au cours de l’histoire évolutive des espèces ou au contraire on été conservés, ainsi que d'établir les relations entre différents traits. Par exemple, des valeurs de traits similaires pour deux espèces ou des corrélations entre traits peuvent être dues à une origine évolutive commune ou au contraire à une convergence évolutive. De fait, des espèces fonctionnellement similaires peuvent appartenir au même clade ou au contraire être phylogénétiquement éloignées. De nombreux outils statistiques ont ainsi été développés pour évaluer la dépendance interspécifique des valeurs de traits dues à la phylogénie. Blomberg (2002) propose ainsi une méthode permettant de détecter et d'évaluer l'importance du « signal phylogénétique » (terme préféré à celui « d'inertie phylogénétique » plus ancien mais dont la signification conceptuelle est plus controversée selon lui) d'un trait au travers d'une phylogénie, c'estàdire la tendance pour des espèces phylogénétiquement proches à plus se ressembler que des espèces éloignées. Pagel (1999) s'est appuyé sur les transformations de longueur de branches pour comparer la vraisemblance d'un continuum de phylogénies modifiées selon différents paramètres et d'évaluer les grandes tendances du patron évolutif du trait. À l'inverse, Ollier (2005) a adopté une démarche se passant des longueurs de branches pour décomposer la variance d'un trait selon une base orthonormée reflétant la topologie de la phylogénie. Moles (2005) s'appuie sur la reconstruction de caractères ancestraux et l'étude de chaque noeud pour déterminer ceux à l'origine de fortes divergences évolutives et leur contribution à la variance actuelle. Enfin, Martins (1997) développa la méthode GLS (generalized least squares) pour remédier à la dépendance phylogénétique des valeurs interspécifiques de traits afin d'étudier les corrélations existant entre différents traits pour mettre en évidence une éventuelle évolution couplée des traits.Le but de ce travail est donc :1 tester, comparer et évaluer les différentes méthodes.
3

2 tester le conservatisme phylogénétique et l'évolution corrélée de deux traits fonctionnels végétaux grâce à ces différentes méthodesAfin de réaliser ces objectifs, on s'intéressera à un échantillon d'espèces localisées de la vallée de la Guisane au col du Galibier (HautesAlpes), zone caractérisée par de forts gradients environnementaux permettant ainsi de travailler sur un spectre large de valeurs de traits. Les données proviennent des campagnes de terrain réalisées par l'équipe TDE (Traits fonctionnels végétaux et Dynamique des Ecosystèmes alpins) du LECA (Laboratoire d'Ecologie Alpine) dont les recherches portent sur le rôle des traits fonctionnels des végétaux dans la structure et la dynamique des écosystèmes alpins, et dans leurs réponses aux forçages environnementaux. Afin de réaliser les objectifs, les données utilisées sont celles collectées dans le cadre de projets précédents (notamment le projet européen VISTA) ainsi que celle effectuée en 2008 dans le cadre du projet Diversitalp financé par l’ANR.On s'intéressera ainsi à deux traits foliaires qui bénéficient d'une attention particulière dans la littérature d'écologie fonctionnelle : l'indice SLA (specific leaf area), calculé comme étant la surface photosynthétique d'une feuille rapportée à sa masse sèche et l'indice LDMC (leaf dry matter content), calculé comme étant la masse sèche de la feuille rapportée à sa masse fraîche. Le SLA est un indice corrélé à la fonction d'acquisition des ressources et au taux de croissance tandis que le LDMC est un trait reflétant la capacité de stockage en matière organique au sein de la feuille et généralement à la durée de vie des feuilles. Ainsi des valeurs de SLA fortes, caractérisent des plantes à croissance rapide produisant de grandes feuilles mais dont la longévité est faible (Westoby 1999) ; de fortes valeurs de LDMC caractérisent des plantes dont les organes foliaires accumulent des nutriments et constituent d'importants pools de stockage donc avec un faible turnover des tissus. Non seulement, ces deux traits reflètent des stratégies écologiques divergentes en tant que traits foliaires reliés à la fonction d'acquisition et stockage des nutriments, mais de plus il est intéressant d'étudier leur relation afin de tester s’il existe une relation de compensation entre ces deux traits et si cette corrélation existe toujours lorsque l’on prend la phylogénie en compte (trade off évolutif).
4
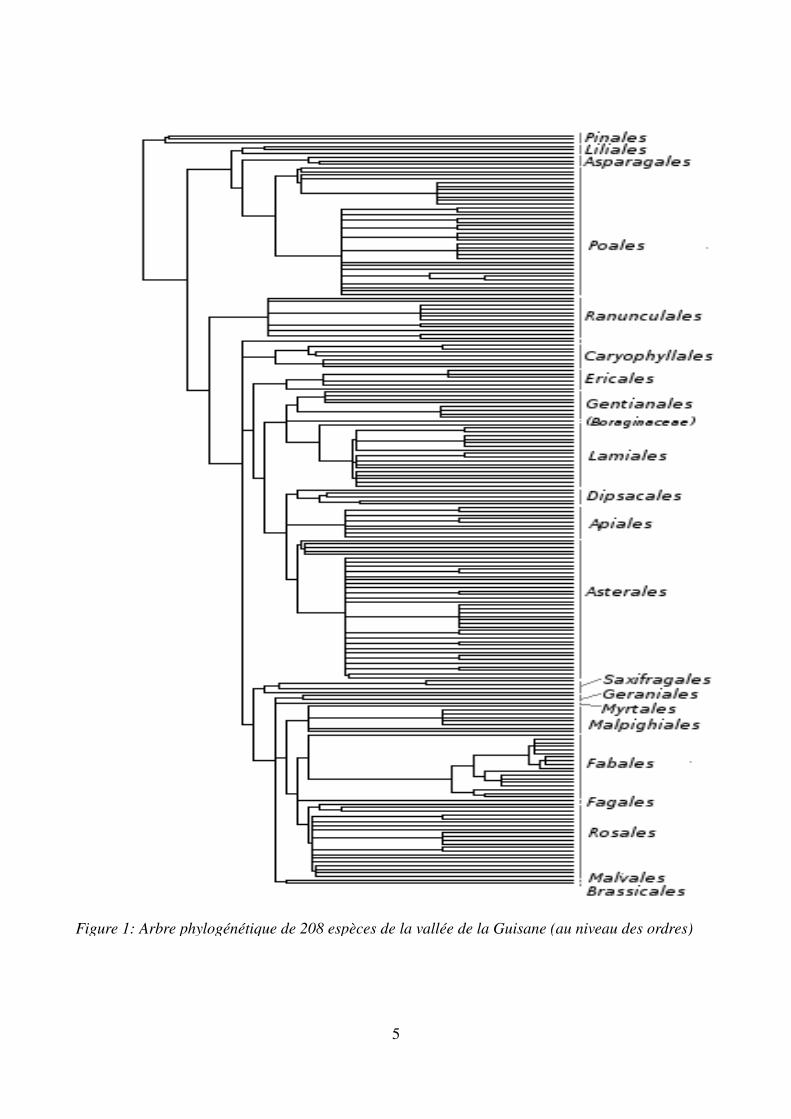
Figure 1: Arbre phylogénétique de 208 espèces de la vallée de la Guisane (au niveau des ordres)
5

Matériel et Méthode
I Acquisition des données sur les traits fonctionnelsLe site d'étude est la vallée de la Guisane (HautesAlpes, France) qui s'étend sur 20 km entre Briançon et le col du Lautaret. Centsept communautés végétales ont été étudiées sur ce site, leur répartition se fait sur un large gradient d'altitude (12962947m). La stratégie d'échantillonnage et de mesures des traits fonctionnels a été élaborée pour compléter les bases de données du LECA déjà existantes de façon à obtenir une représentativité totale de 80% de couverture végétale. La campagne Diversitalp de l'été 2008 a ainsi échantillonné cent espèces végétales.Douze individus de chaque espèce ont été échantillonnés sur le terrain et conservé en milieu frais et humide afin de les préserver de la dessiccation. Les individus collectés étaient séparés de plusieurs mètres sur le site afin d'éviter d'échantillonner des clones ou des individus génétiquement proches, ainsi que des individus présents dans des conditions microenvironnementales similaires (limitation de la pseudoréplication dans nos données).Les traits foliaires SLA (specific leaf area) et LDMC (leaf dry matter content) étaient ensuite estimés, à partir de la masse fraîche, de la surface foliaire mesurée en laboratoire par scanner HP grâce au logiciel MideBMP (entre 0 et 5% d'erreur) et la masse sèche foliaire mesurée sur une balance de précision (microgramme) après quatre jours à l’étuve (60°C).
II Analyse phylogénétique
1. ArbreL'analyse a été effectuée à partir d'un superarbre (Figure 1) restreint aux espèces contenues dans la base de données et dont à la fois les valeurs de SLA et de LDMC étaient disponibles. L'arbre phylogénétique comporte ainsi 208 feuilles. De nombreux embranchements, notamment au niveau des genres ne sont pas résolus et sont donc polytomiques.L'arbre phylogénétique a été construit grâce à la compilation de phylogénies (approche du « superarbre » réalisées par différents partenaires du projet DIVERSITALP. L'élaboration de ce superarbre n'est pas le sujet de ce travail, ainsi je n'ai pas participé à son élaboration.
2. Outils statistiquesDifférentes méthodes ont été utilisées afin d'analyser les données de traits d'un point de vue phylogénétique. Toutes ont été appliquées grâce au logiciel R (Ihaca, R, Gentleman R., 1996. R : a language for data analysis and graphics. Journal of Computational and Graphical Statistics 5 299314)
1. Le et (Pagel 1999, Verdù 2006) :
La démarche consiste à appliquer différents modèles aux données phylogénétiques en faisant varier le paramètre concerné. Ce paramètre modifie les longueurs de branches pour arriver au modèle le plus vraisemblant*, c'estàdire celui prédisant au mieux le patron évolutif du trait..Le paramètre est simil *aire à l'indice développé par Blomberg (2002) pour détecter un signal phylogénétique, c'estàdire la tendance pour des taxons proches d'avoir des caractères proches. Une
* cf. Glossaire
6

valeur de nulle indique une absence de signal phylogénétique, cela indique que le trait a évolué comme sur une phylogénie en étoile (toutes les espèces sont équidistances entre elles, sans relations phylogénétiques). Une valeur de égale à 1 indique que le trait évalue exactement tel que la topologie originale de la phylogénie le suggérait.Le mode de l'évolution est mesurée par la comparaison des vraisemblances de modèles avec différentes valeurs de . Ce paramètre raccourcit ou étire individuellement les branches de l'arbre phylogénétique ; ainsi une valeur de supérieure à 1 indique que les longues branches ont besoin d'être plus étirées que les branches courtes, traduisant une accélération de l'évolution du trait le long des plus longues branches. . Une valeur inférieure à 1 traduisant plutôt une conservation du trait sur les branches plus longues (donc trait plutôt statique). Une valeur de nulle indique un cas de ponctualisme, c'estàdire un trait dont le changement est rapide sur une courte période de temps (évolution par paliers), tandis qu'une valeur de égale à 1 indique un cas de gradualisme, c'estàdire un trait dont la vitesse de changement est constante au cours du temps.Le tempo de l'évolution du caractère est mesuré par . Ce paramètre représente la puissance à laquelle sont élevées les chemins évolutifs (distance de la racine aux feuilles). Si ce paramètre est inférieur à 1, cela signifie que les chemins évolutifs courts contribuent plus à l'évolution du caractère que les chemins longs. Ce qui est cohérent avec des cas de divergences précoces du trait. Inversement, une valeur de supérieure à 1 traduit une radiation tardive du trait.
Les vraisemblances associées à ces trois modèles ont été comparées à celles de modèles pour des valeurs de paramètre ( , fixées à 0 ou à 1, ces valeurs de paramètre correspondant à des « valeursfrontière » (cf. paragraphe précédent). Les pvalues associées ont ensuite été calculées par approximation du ratio de vraisemblance* par la loi de chi2 à un degré de liberté*. Le script associé est disponible en annexe. Le script utilise notamment la fonction fitContinuous contenue dans la librairie R « geiger »
2. Orthogramme (Ollier et al. 2005)
Cette méthode consiste à traduire la topologie de l'arbre phylogénétique par une matrice et de déduire la base orthornomée associée.. Il convient de noter que la démarche ne prend pas en compte les longueurs de branches de la phylogénie et se focalise uniquement sur les noeuds (topologie).Au final, la base orthonormée résume ainsi chaque noeud de l'arbre par un ou plusieurs (dans le cas de polytomies) vecteurs de la base orthonormée (cf. Figure 1).La décomposition de la variance du trait selon les vecteurs de la base orthonormée permet donc de calculer la contribution de chaque vecteur de la base à la variabilité actuelle du trait. Ces contributions sont comparées à un modèle nul (par randomisation des feuilles de l'arbre phylogénétique).
La fonction orthogram() disponible sous R au sein de la librairie ade4, fournit également quelques
* Cf. Glossaire
7
Figure 2: Représentation des vecteurs de la base orthonormée pour un arbre fictif. Les vecteurs Bi
sont ceux de la base orthonormée et Li, ceux associés à chacune des feuilles de l'arbre. (Ollier et al. 2005)

outils permettant d'interpréter cette décomposition. Elle renvoit l'orthogramme proprement dit, c'estàdire la contribution de chaque vecteur à la variance du trait, ainsi que l'orthogramme cumulé. De plus, quatre indices sont calculés :Soit ri la composante du vecteur « trait » centré et standardisé (à partir du vecteur x contenant les valeurs de traits pour chacune des feuilles) le long du vecteur i (compris entre 1 et (t1) où t est le nombre de feuilles de l'arbre phylogénétique) de la base orthonormée.
R2max x =max r 12 ,. . . ,r t−1
2 Cet indice permet de constater si un vecteur de la base contribue dans une large mesure à la variance totale, c'estàdire qu'un noeud de l'arbre contribue beaucoup à la variance du trait, ce dernier étant ensuite conservé dans les branches descendantes.
∑i= 1
m
ri2−
mt−1
Cet indice permet de constater une dépendance plus diffuse de la variance du trait dans l'arbre phylogénétique en calculant l'écart maximum de l'orthogramme cumulatif à la droite d'évolution moyenne.
SkR2k x =∑i= 1
t−1
ir i2
Cet indice permet de calculer une distribution de la variance plutôt au niveau de la racine ou au niveau des feuilles, c'estàdire de constater une divergence plutôt précoce ou tardive.
SCE x =∑i=1
t−1
r i2−r i−1
2 Cet indice permet de calculer la variation locale moyenne des valeurs de l'orthogramme.
3. Divergence et contribution des noeuds (Moles et al. 2005) :
Un script (disponible en annexe) destiné à être utilisé sur R a été codé pour intégralement reprendre cette analyse. Moles passe par une estimation des caractères ancestraux utilisant la méthode des contrastes phylogénétiquement indépendants (PIC) de Felsenstein (1985), néanmoins cette étude lui a préféré la méthode GLS (generalized least squares, Martins et Hansen 1997) s'appuyant sur une hypothèse d'évolution brownienne* (ce qui revient à la méthode PIC). Ce choix s'est justifié par la comparaison de plusieurs méthodes d'évaluation *des caractères ancestraux et la constatation de la présence d'un outlayer* dans le set de caractères ancestraux évalué par la méthode PIC.De plus, le problème des polytomies* (que la méthode ne prend pas en charge) a été résolu par l'ajout d'embranchements aléatoires permettant d'obtenir un arbre phylogénétique entièrement dichotomique. Les branches supplémentaires sont alors de longueurs nulles. Afin d'évaluer l'impact de cette transformation sur la valeur de caractère ancestral attribuée à un noeud polytomique, un arbre polytomique (39 espèces et 9 noeuds internes)a été résolu cent fois en arbre dichotomique et la valeur du caractère ancestral de la racine a été évaluée. Cette dernière reste identique. On peut en déduire que cet ajout d'embranchement ne modifie pas l'évaluation du caractère ancestral, ce qui peut, par ailleurs, s'expliquer par la nullité des branches ajoutées.Par la suite, l'analyse a été conduite à partir de l'arbre original sans tenir compte des noeuds ajoutés pour les besoins du calcul des états ancestraux afin de ne pas sousestimer l'importance des noeuds polytomiques dans le calcul de leur divergence et leur contribution
L'amplitude de divergence a ensuite été calculée pour chaque noeud, il s'agit de la racine carrée de
* Cf. Glossaire
8

la somme des différences entre les valeurs de caractères des noeuds fils et du noeud père.La contribution de chaque noeud a également été déterminée, cet indice permet d'estimer la contribution du noeud à la variance actuelle du trait dans tout l'arbre. Les détails des calculs sont disponibles en annexe.Afin d'évaluer leur significativité, ces résultats ont été comparés avec ceux obtenus par 999 randomisations des feuilles de l'arbre phylogénétique. Cela constitue également une différence avec la démarche de Moles qui n'utilise ces randomisations que pour l'étude des amplitudes de divergence (voir discussion)Afin de pouvoir comparer les valeurs d'amplitude de divergence et de contribution, pour chaque noeud, la moyenne des contributions et des amplitudes de divergences obtenues lors des randomisations a été calculée. « L'anomalie » de divergence et de contribution a ensuite été calculée en soustrayant cette moyenne aux valeurs observées de divergence et de contribution.
4. Phylogenetic least squares model (Martins et Hansen 1997 ; RossIbarra 2007) ou PGLS
Cette méthode permet, entre autres, de calculer la corrélation entre deux traits en prenant en compte des relations phylogénétiques qui relient les espèces. De plus, le modèle proposé par Martins permet d'évaluer le paramètre qui reflète la contrainte évolutive. Une valeur nulle illustre une croissance linéaire de la divergence phénotypique entre espèces avec la distance phylogénétique et revient à une hypothèse d'évolution brownienne (divergence aléatoire ou pression de sélection variable) et à la méthode PIC de Felsenstein. Un GLS avec une valeur de non nulle fait l'hypothèse d'une croissance exponentielle de la divergence phénotypique avec la distance phylogénétique. Le modèle GLS converge vers un modèle classique de corrélation entre valeurs indépendantes quand tend vers l'infini. Cette démarche a été effectuée avec le logiciel Compare (Martins 2004).
9
Résultats
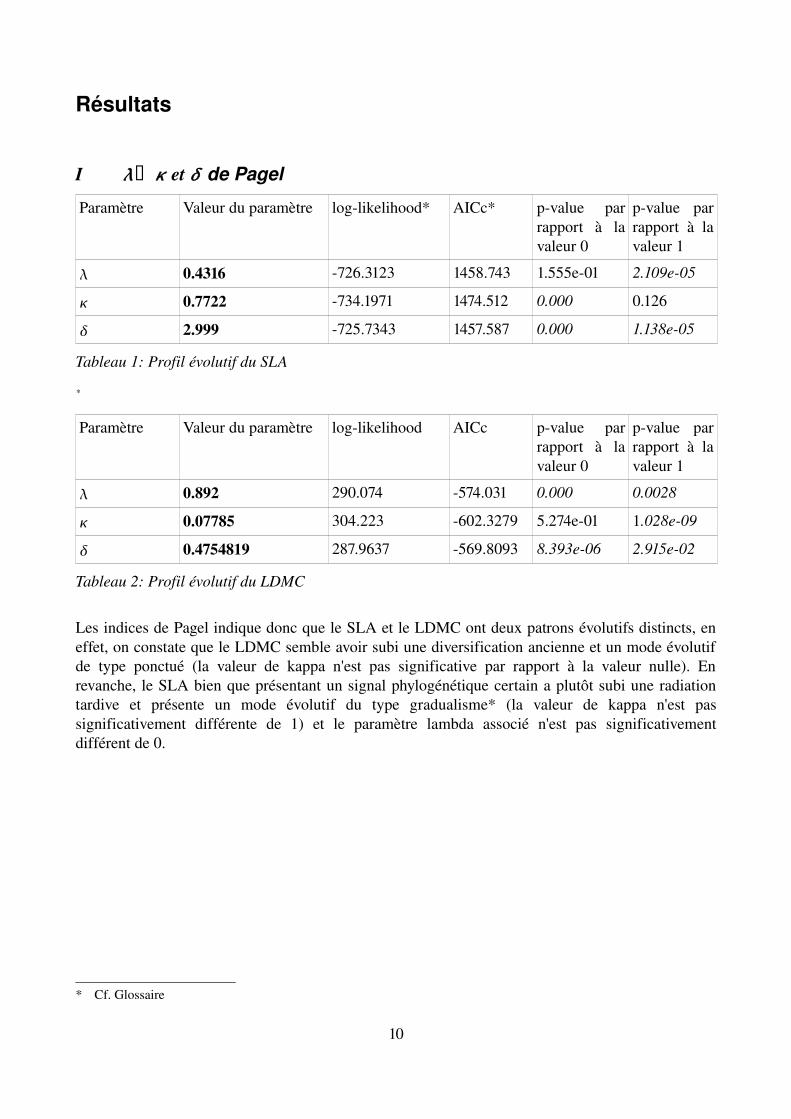
I x et de Pagel
Paramètre Valeur du paramètre loglikelihood* AICc* pvalue par rapport à la valeur 0
pvalue par rapport à la valeur 1
0.4316 726.3123 1458.743 1.555e01 2.109e05
0.7722 734.1971 1474.512 0.000 0.126
2.999 725.7343 1457.587 0.000 1.138e05
Tableau 1: Profil évolutif du SLA
*
Paramètre Valeur du paramètre loglikelihood AICc pvalue par rapport à la valeur 0
pvalue par rapport à la valeur 1
0.892 290.074 574.031 0.000 0.0028
0.07785 304.223 602.3279 5.274e01 1.028e09
0.4754819 287.9637 569.8093 8.393e06 2.915e02
Tableau 2: Profil évolutif du LDMC
Les indices de Pagel indique donc que le SLA et le LDMC ont deux patrons évolutifs distincts, en effet, on constate que le LDMC semble avoir subi une diversification ancienne et un mode évolutif de type ponctué (la valeur de kappa n'est pas significative par rapport à la valeur nulle). En revanche, le SLA bien que présentant un signal phylogénétique certain a plutôt subi une radiation tardive et présente un mode évolutif du type gradualisme* (la valeur de kappa n'est pas significativement différente de 1) et le paramètre lambda associé n'est pas significativement différent de 0.
* Cf. Glossaire
10
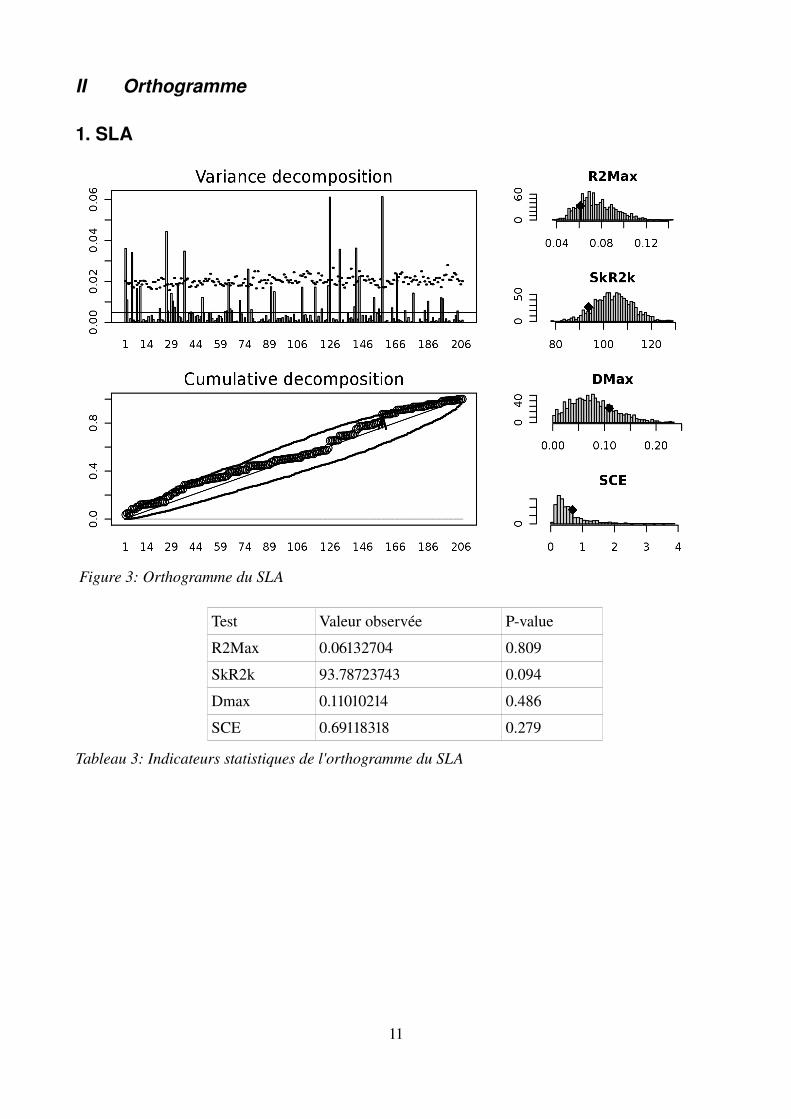
II Orthogramme
1. SLA
Test Valeur observée Pvalue
R2Max 0.06132704 0.809
SkR2k 93.78723743 0.094
Dmax 0.11010214 0.486
SCE 0.69118318 0.279
Tableau 3: Indicateurs statistiques de l'orthogramme du SLA
11
Figure 3: Orthogramme du SLA
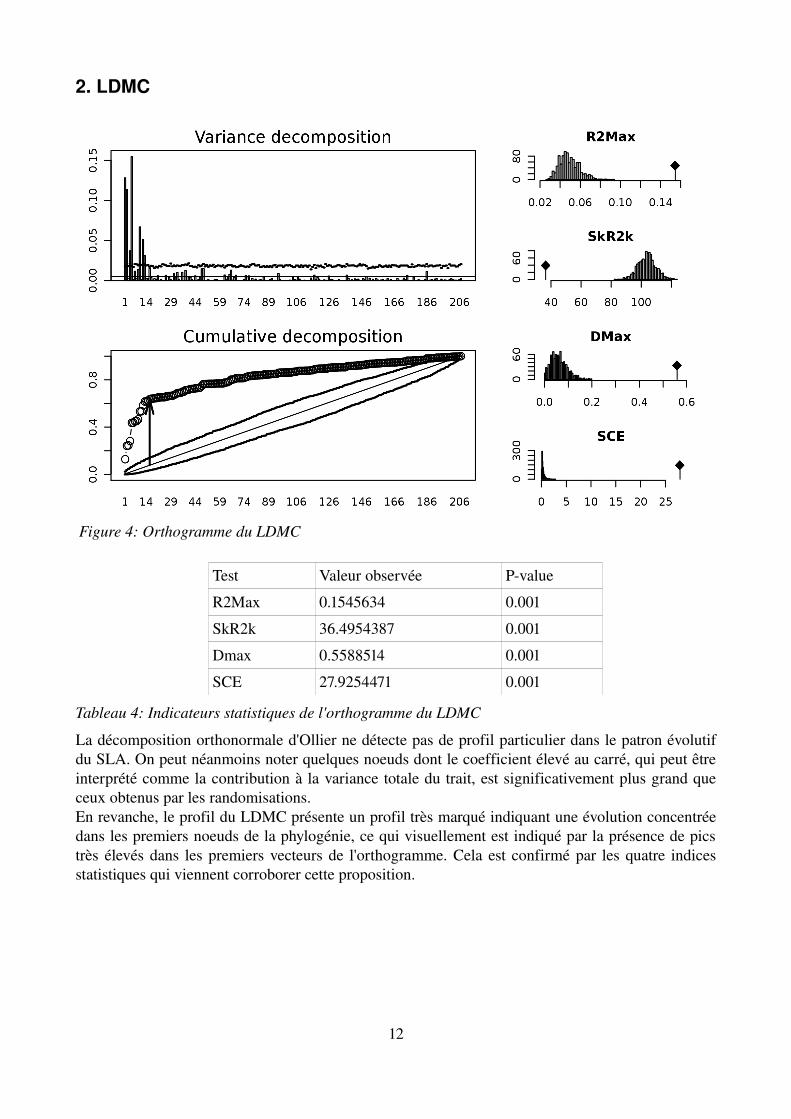
2. LDMC
Test Valeur observée Pvalue
R2Max 0.1545634 0.001
SkR2k 36.4954387 0.001
Dmax 0.5588514 0.001
SCE 27.9254471 0.001
Tableau 4: Indicateurs statistiques de l'orthogramme du LDMC
La décomposition orthonormale d'Ollier ne détecte pas de profil particulier dans le patron évolutif du SLA. On peut néanmoins noter quelques noeuds dont le coefficient élevé au carré, qui peut être interprété comme la contribution à la variance totale du trait, est significativement plus grand que ceux obtenus par les randomisations.En revanche, le profil du LDMC présente un profil très marqué indiquant une évolution concentrée dans les premiers noeuds de la phylogénie, ce qui visuellement est indiqué par la présence de pics très élevés dans les premiers vecteurs de l'orthogramme. Cela est confirmé par les quatre indices statistiques qui viennent corroborer cette proposition.
12
Figure 4: Orthogramme du LDMC
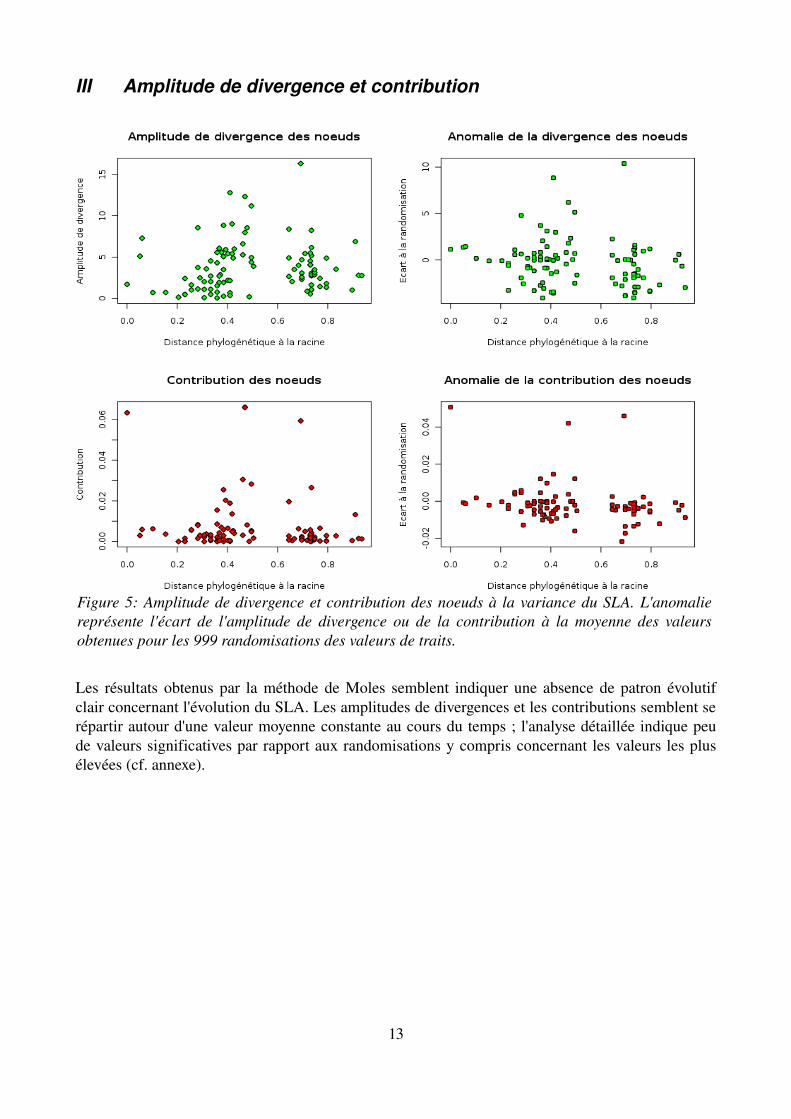
III Amplitude de divergence et contribution
Les résultats obtenus par la méthode de Moles semblent indiquer une absence de patron évolutif clair concernant l'évolution du SLA. Les amplitudes de divergences et les contributions semblent se répartir autour d'une valeur moyenne constante au cours du temps ; l'analyse détaillée indique peu de valeurs significatives par rapport aux randomisations y compris concernant les valeurs les plus élevées (cf. annexe).
13
Figure 5: Amplitude de divergence et contribution des noeuds à la variance du SLA. L'anomalie représente l'écart de l'amplitude de divergence ou de la contribution à la moyenne des valeurs obtenues pour les 999 randomisations des valeurs de traits.
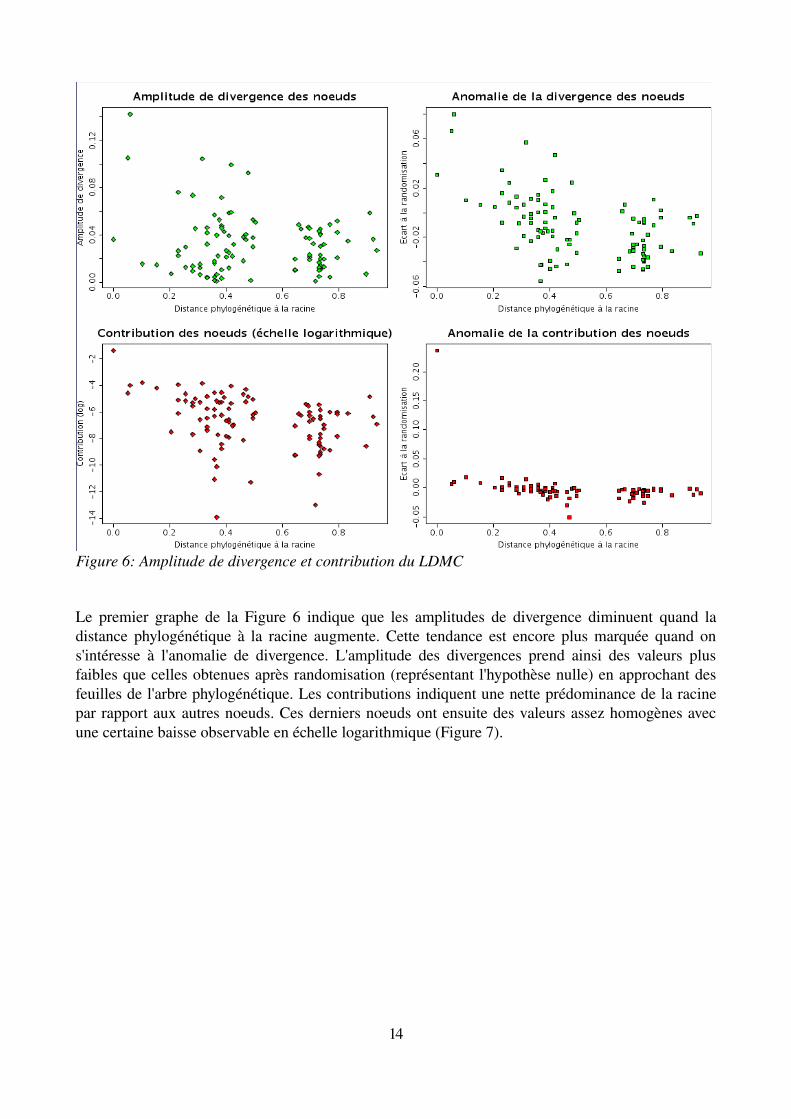
Le premier graphe de la Figure 6 indique que les amplitudes de divergence diminuent quand la distance phylogénétique à la racine augmente. Cette tendance est encore plus marquée quand on s'intéresse à l'anomalie de divergence. L'amplitude des divergences prend ainsi des valeurs plus faibles que celles obtenues après randomisation (représentant l'hypothèse nulle) en approchant des feuilles de l'arbre phylogénétique. Les contributions indiquent une nette prédominance de la racine par rapport aux autres noeuds. Ces derniers noeuds ont ensuite des valeurs assez homogènes avec une certaine baisse observable en échelle logarithmique (Figure 7).
14
Figure 6: Amplitude de divergence et contribution du LDMC
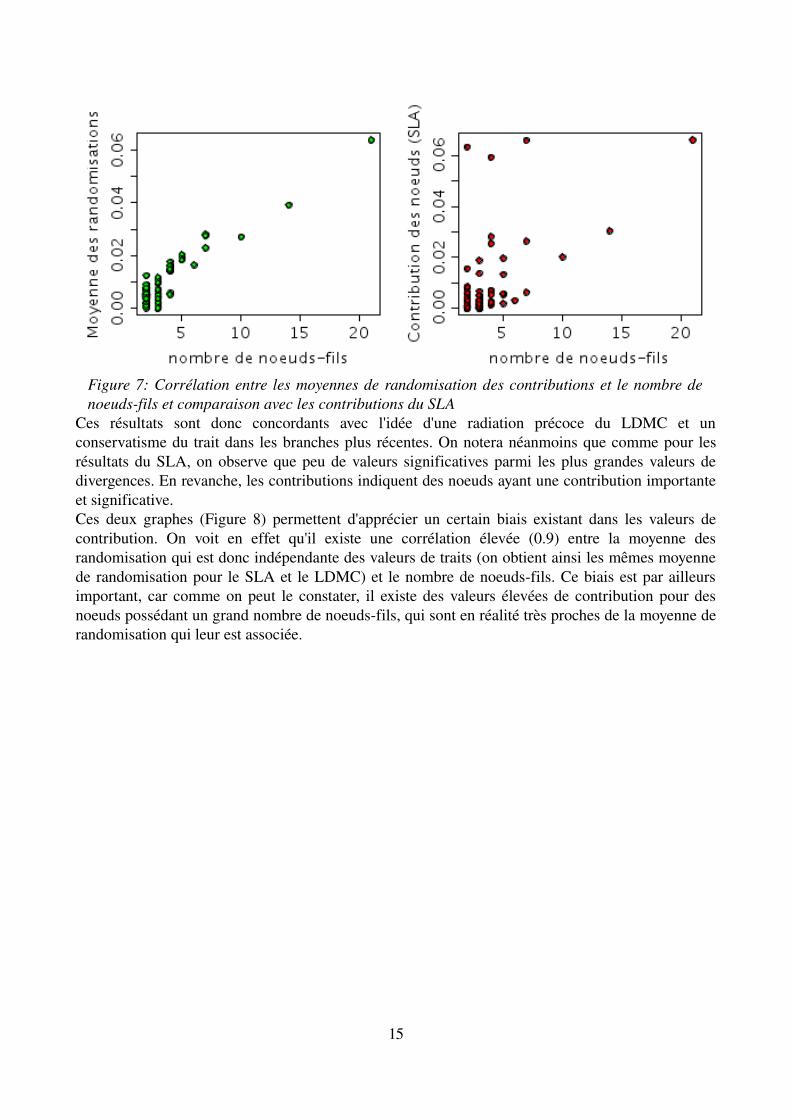
Ces résultats sont donc concordants avec l'idée d'une radiation précoce du LDMC et un conservatisme du trait dans les branches plus récentes. On notera néanmoins que comme pour les résultats du SLA, on observe que peu de valeurs significatives parmi les plus grandes valeurs de divergences. En revanche, les contributions indiquent des noeuds ayant une contribution importante et significative.Ces deux graphes (Figure 8) permettent d'apprécier un certain biais existant dans les valeurs de contribution. On voit en effet qu'il existe une corrélation élevée (0.9) entre la moyenne des randomisation qui est donc indépendante des valeurs de traits (on obtient ainsi les mêmes moyenne de randomisation pour le SLA et le LDMC) et le nombre de noeudsfils. Ce biais est par ailleurs important, car comme on peut le constater, il existe des valeurs élevées de contribution pour des noeuds possédant un grand nombre de noeudsfils, qui sont en réalité très proches de la moyenne de randomisation qui leur est associée.
15
Figure 7: Corrélation entre les moyennes de randomisation des contributions et le nombre de noeudsfils et comparaison avec les contributions du SLA
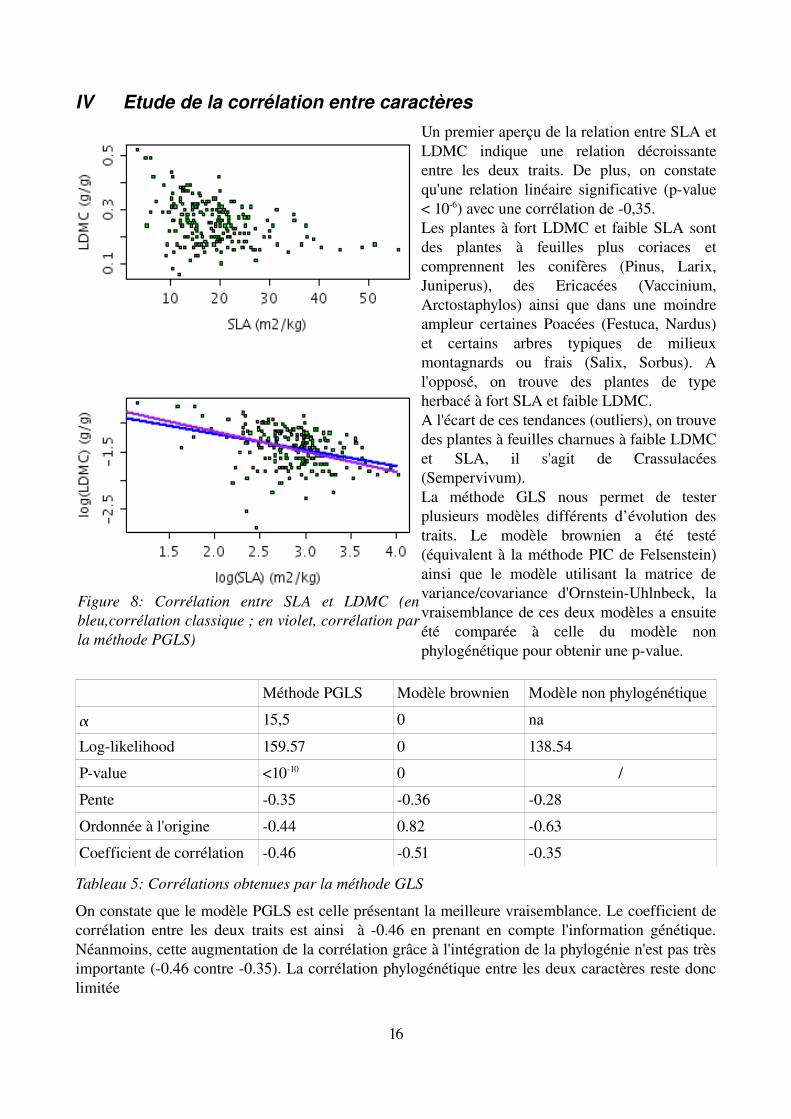
IV Etude de la corrélation entre caractèresUn premier aperçu de la relation entre SLA et LDMC indique une relation décroissante entre les deux traits. De plus, on constate qu'une relation linéaire significative (pvalue < 106) avec une corrélation de 0,35.Les plantes à fort LDMC et faible SLA sont des plantes à feuilles plus coriaces et comprennent les conifères (Pinus, Larix, Juniperus), des Ericacées (Vaccinium, Arctostaphylos) ainsi que dans une moindre ampleur certaines Poacées (Festuca, Nardus) et certains arbres typiques de milieux montagnards ou frais (Salix, Sorbus). A l'opposé, on trouve des plantes de type herbacé à fort SLA et faible LDMC. A l'écart de ces tendances (outliers), on trouve des plantes à feuilles charnues à faible LDMC et SLA, il s'agit de Crassulacées (Sempervivum). La méthode GLS nous permet de tester plusieurs modèles différents d’évolution des traits. Le modèle brownien a été testé (équivalent à la méthode PIC de Felsenstein) ainsi que le modèle utilisant la matrice de variance/covariance d'OrnsteinUhlnbeck, la vraisemblance de ces deux modèles a ensuite été comparée à celle du modèle non phylogénétique pour obtenir une pvalue.
Méthode PGLS Modèle brownien Modèle non phylogénétique
15,5 0 na
Loglikelihood 159.57 0 138.54
Pvalue <1010 0 /
Pente 0.35 0.36 0.28
Ordonnée à l'origine 0.44 0.82 0.63
Coefficient de corrélation 0.46 0.51 0.35
Tableau 5: Corrélations obtenues par la méthode GLS
On constate que le modèle PGLS est celle présentant la meilleure vraisemblance. Le coefficient de corrélation entre les deux traits est ainsi à 0.46 en prenant en compte l'information génétique. Néanmoins, cette augmentation de la corrélation grâce à l'intégration de la phylogénie n'est pas très importante (0.46 contre 0.35). La corrélation phylogénétique entre les deux caractères reste donc limitée
16
Figure 8: Corrélation entre SLA et LDMC (en bleu,corrélation classique ; en violet, corrélation par la méthode PGLS)

Discussion
Patron évolutif du SLA et du LDMCLes différentes méthodes employées fournissent des réponses cohérentes. Il est ainsi net que le patron évolutif que semble suivre le LDMC est celui d'une radiation précoce (Tableau 2, Tableau 4 Figure 6). Cela entraîne un fort signal phylogénétique (Tableau 2)étant donné le fort conservatisme phylogénétique de ce trait dans les noeuds au delà des premières divergences. Cela se manifeste par les faibles valeurs d'amplitude de divergence des noeuds les plus récents, fait qui se retrouve aussi bien dans la méthode de Moles que d'Ollier. De même, les trois méthodes semblent montrer que le SLA a un profil évolutif peu tranché. Les méthodes de Moles et d'Ollier n'indiquent aucune tendance globale nette. La méthode de Pagel indique un signal phylogénétique moyen ainsi qu'un indice élevé. Cela signifie que le trait a subi une radiation tardive et donc un conservatisme phylogénétique faible, ce qui n'est en revanche pas détectée par les deux autres méthodes. La méthodes de Moles indiquent des contributions importantes de certains noeuds sans pour autant qu'il y ait de corrélation particulière avec la distance phylogénétique à la racine. Le profil de l'orthogramme d'Ollier s'interprète comme un cas où la dépendance phylogénétique n'est pas significative, ce qui est confirmé avec la valeur du paramètre , non significativement différente de 0.Ceci indique un moins fort conservatisme phylogénétique. Le SLA semble donc être un trait fonctionnel plus labile et moins contraint par la sélection naturelle que le LDMC.
Corrélation des deux patrons d'évolutionLes deux traits présentent une corrélation significative qui indique que ces deux caractères sont liés, ce qui n'est guère étonnant étant donné qu'ils se rapportent tout deux au même organe végétatif. Les plantes à fort SLA possèdent un faible LDMC et inversement. On remarque que l'ajout de la contrainte phylogénétique par le biais du modèle d'OrnsteinUhlnbeck améliore le coefficient de corrélation du modèle linéaire ce qui confirme la tendance globale que l'on a pu observer avec un modèle linéaire classique. Le modèle exponentiel se révèle plus approprié, ce qui concorde avec les conclusions précédentes indiquant que l'évolution des traits est soumis à des contraintes évolutives. Cela est cohérent avec les stratégies écologiques que reflètent ces traits fonctionnels. En effet, les végétaux à fort SLA et faible LDMC sont des plantes donc la croissance sera rapide et l'acquisition des ressources efficace à court terme par rapport à la matière organique investie. En revanche, les structures foliaires ont une moins grande longévité car plus fragiles (plus sensibles à la dessiccation par exemple). A l'inverse, les plantes à fort LDMC et faible SLA, présentent une croissance moins rapide mais des feuilles à plus grande longévité, ce qui permet à ces feuilles de stocker des nutriments notamment l'azote. (Westoby 1988). On remarque également que le paramètre est élevé, ce qui indique que l'autocorrélation phylogénétique donnant le modèle le plus vraisemblable n'est pas très importante, ce qui peut explique l'augmentation modérée de la valeur absolue du coefficient de corrélation. On peut relier cette faible autocorrélation au fait que les deux patrons évolutifs sont globalement découplés et que les fortes divergences évolutives ne correspondent pas.
Critique de la démarche de MolesL'article de Moles indique l'utilisation de randomisations des valeurs de traits pour évaluer la significativité des résultats. Il apparaît que l'étude iciprésente ne met en valeur que peu de valeurs
17

significatives (cf. Annexe) même parmi les noeuds les plus importants. Or une valeur de contribution ou d'amplitude de divergence même élevée et non significative indique qu'en moyenne, même après randomisation des valeurs et donc perte du lien phylogénétique entre cellesci, ce noeud possède encore une contribution ou une amplitude de divergence élevée ; ceci est d'autant plus gênant que l'analyse de Moles met en valeur les noeuds possédant les contributions ou divergences les plus élevées. On peut alors se demander si les indices de contribution et d'amplitude de divergence possèdent un biais qui les rend trop dépendants de la topologie de l'arbre phylogénétique par rapport aux valeurs de traits attribués aux feuilles. Il apparaît en effet que la moyenne des randomisations des contribution est variable selon le noeud (contrairement à ce que donne la méthode d'Ollier) et se corrèle très bien au nombre de noeudsfils du noeud étudié (Figure 8), ce qui n'est pas surprenant, compte tenu de la formule de l'indice de contribution. Ce biais est donc particulièrement important pour des arbres comprenant de nombreuses polytomies comme celui étudié ici. Ceci justifie l'usage de « l'anomalie » en particulier pour l'indice de contribution.
De plus, la démarche de Moles semble peu adaptée à l'étude de traits fonctionnels tel que le LDMC qui présente dans la phylogénie étudiée ici, un patron évolutif très marqué. En effet, l'estimation des caractères ancestraux reposent sur l'hypothèse d'une évolution brownienne du trait (Felsenstein 1985) ; hors ce modèle semble totalement inapproprié pour un trait tel que le LDMC dont le profil n'est clairement pas celui d'une évolution brownienne.Si on se reste dans le cadre d'étude proposé par Moles (reconstruction de caractères ancestraux), il pourrait être intéressant de comparer les résultats obtenus pour différents méthodes de calcul de caractères ancestraux notamment prenant en compte une sélection évolutive. Néanmoins, il n'existe que peu de méthodes de reconstruction de caractères ancestraux adaptés à cela.
Comparaison des trois méthodesLes trois méthodes présentent des stratégies différentes. La méthode de Pagel s'appuie sur l'étude de la vraisemblance de différents modèles s'appuyant sur la modifications des longueurs de branches pour déterminer des profils évolutifs sur l'ensemble de l'arbre phylogénétique. Elle permet notamment également de quantifier le mode évolutif ainsi que le « signal phylogénétique » en plus du tempo. La démarche d'Ollier et de Moles relèvent elles d'une analyse statistique qui permet de tirer des informations sur chacun des noeuds. L'orthogramme d'Ollier présente la particularité de ne s'appuyer que sur les noeuds. Cela permet de se détacher de l'information parfois peu fiable selon l'auteur que donnent les longueurs de branches en particulier pour des superarbres, cela conduit néanmoins à une perte d'information. Ainsi l'étude d'arbres phylogénétiques présentant des longueurs de branches contrastées conduit à des résultats contradictoires entre la méthode d'Ollier et de Pagel. Enfin la méthode de Moles s'appuie, elle, sur l'estimation de caractères ancestraux pour déterminer l'amplitude de divergence et la contribution de chaque noeud. Néanmoins, il apparaît que l'indice de contribution possède un biais intrinsèque qui conduit à sa surévaluation pour des noeuds polytomiques ce qui peut s'avérer gênant pour des arbres possédant des très fortes polytomies dues à une nonrésolution de certains embranchements. L'usage de « l'anomalie », c'estàdire de la différence entre la valeur de contribution ou de divergence et la moyenne obtenue par les randomisations (qui traduit le biais phylogénétique) permet de rendre plus comparables les valeurs attribuées aux différents noeuds.
Il est finalement intéressant de constater que les grandes tendances données par ces trois méthodes
18

ne se contredisent pas et présentent des résultats complémentaires quant au profil évolutif des traits. On constate néanmoins que si les méthodes d'Ollier et de Moles fournissent des résultats simlaires au niveau de l'arbre entier, ce n'est pas tout à fait le cas au niveau des noeuds (cf. Annexe).Au final, il paraît donc risqué dans cette étude de raisonner au niveau des noeuds et de mettre en valeur ceux censés refléter les grandes divergences des patrons évolutifs du SLA et du LDMC comme cela a été fait dans l'étude faite par Moles sur la masse des graines. Ceci est d'autant plus vrai que l'arbre ici présente un set limité d'espèces, rendant notamment les valeurs de contribution et de divergences peu fiables au niveau des noeuds tardifs.
ConclusionL'utilisation de trois méthodes statistiques différentes a permis de comparer leurs résultats et d'apprécier leurs qualités respectives et d'avoir une idée de leurs limites. Ainsi l'étude conduit à indiquer que le LDMC est un trait très conservé et que sa radiation est précoce. Les indices de Pagel indique ainsi un fort signal phylogénétique qui montre que le déterminisme phylogénétique est très important. A l'inverse, l'indice SLA semble plus labile, sa radiation plus tardive et le profil donné par les indices de Pagel est comparable à celui d'une évolution brownienne. A ce titre, ce trait semble moins conservé. Le LDMC est donc un trait plus déterminé que le SLA par son histoire évolutive. Ce conservatisme phylogénétique peut être expliqué par l'existence de contraintes évolutives (physiologiques, génétiques...) limitant les possibilités de changement du LDMC. On peut comparer cela à une échelle intraspécifique, où on observe une plus grande variabilité du SLA entre individus, par exemple pour des plantes subissant un phénomène d'étiolement, que pour le LDMC plus constant. Malgré ces profils évolutifs différents, les deux traits présentent une corrélation significative (0.46 en prenant en compte l'autocorrélation phylogénétique par ailleurs, peu importante), ce qui indique la nécessité de la présence d'autres traits expliquant que le SLA et le LDMC n'évoluent pas ensemble, par exemple l'épaisseur des feuilles. Par ailleurs, l'utilisation et la comparaison des résultats données par les méthodes de Pagel (1999), Ollier (2005) et Moles (2005) a permis de mettre en lumière leurs limites et leurs qualités respectives. Ainsi, la méthode de Pagel s'avère intéressante pour évaluer différents paramètres se référant à l'ensemble de l'arbre phylogénétique mais ne permet pas de s'intéresser aux différences entre embranchements. En se focalisant sur les noeuds, les démarches d'Ollier et Moles permettent de voir précisément où se situent les grandes radiations évolutives. Néanmoins, leurs résultats ne correspondent pas tout à fait et cela nous a permis de mettre en lumière un biais existant dans la méthode de Moles. Il serait par la suite intéressant d'approfondir, notamment en généralisant la démarche d'étude, l'importance de ce biais afin d'avoir un regard critique sur la méthode statistique.
19

BibliographieBlomberg S.P., Garland T., Tempo and mode in evolution: phylogenetic inertia, adaptationand comparative methods, J. Evol. Biol. 15 (2002)
Díaz S. et al., The plant traits that drive ecosystems: Evidence from three continents, Journal of vegetation science 15 (2004)
Díaz S. et Cabido M., Vive la différence: plant functional diversity matters to ecosystem processes; Trends in Ecology & Evolution 16 (2001)
Felsenstein, J. 1985. Phylogenies and the comparative method; The American Naturalist 125 (1985)
Martins E., Hansen T.; Phylogenies and the comparative method: a general approach to incorporating phylogenetic information into the analysis of interspecific data; The American Naturalist 149 (1997)
Martins, E. 2004). COMPARE. Computer Programs for the Statistical Analysis of Comparative Data. Logiciel mis à disposition par l'auteur sur http://compare.bio.indiana.edu/.
Moles A., Ackerly D., Webb C., Tweddle J.,Dickie J., Westoby M., A brief history of seed size, Nature 307 (2005)
Ollier S., Couteron P., Chessel D., Orthonormal Transform to Decompose the Variance of a LifeHistory Trait Across a Phylogenetic Tree, Biometrics (2005)
Pagel M, Inferring the historical patterns of biological evolution; Nature 401 (1999)
RossIbarra J., Genome size and recombination in angiosperms: a second look, J. Evol. Biol. (2007)
Verdù M., Tempo, mode and phylogenetic associations of relative embryo size evolution in angiosperms, J. Evol Biol. 19 (2006)
Violle C., Navas ML., Vile D., Kazakou E., Fortunel C., Hummel I, Garnier E; Let the concept of trait be functional! Oikos 116 (2007)
Westoby M., A leafheightseed (LHS) plant ecology strategy scheme, Plant and Soil 199 (1998)
20

GlossaireAICc : (Akaike's information criterion), cette mesure permet d'évaluer l'adéquation d'un modèle statistique à un jeu de données. Sa formule est : AIC = 2*(nombre de paramètre)ln(Vraisemblance optimale). Cet indice prend ainsi en compte le nombre de paramètres que comprend le modèle en plus de la vraisemblance. En effet, un modèle statistique perd de sa force avec le nombre de paramètres utilisé pour décrire le système. Un modèle est donc optimal pour une valeur de AIC minimale qui cherche à trouver le modèle optimal décrivant au mieux le système pour un minimum de paramètres libres.L'AICc est l'AIC pourvu d'une correction permettant de l'appliquer à des échantillons de taille réduite (l'AICc converge vers l'AIC pour de larges échantillons).
Evolution brownienne : évolution hypothétique où le trait varie au hasard. Pour un trait de variance 2 , la loi de probabilité de la valeur du trait à t+dt est une loi normale de moyenne égale à la valeur du trait à l'instant t et de variance 2.
Outlayer : Valeur extrême par rapport au reste de l'échantillon.
Polytomies : Qualifie un noeud ayant plus de deux descendants. On distingue les polytomies « soft » qui sont des noeuds dont la configuration n'est pas résolues et les polytomies « hard » qui sont résolues et sont donc des divergences entre plus de deux branches.
Vraisemblance/likelihood : fonction permettant de décrire la pertinence d'un modèle statistique à un échantillon. Le maximum de vraisemblance est atteint pour un set de paramètres permettant le meilleur fit du modèle avec l'échantillon. Plus le maximum de vraisemblance est important, plus le modèle est pertinent pour décrire l'échantillon.
21

Annexes
Annexe 1 : Démarche mathématique de Moles
1. Estimation des états ancestraux (Felsenstein 1985)Soit Xk, la valeur de caractère ancestral du noeud en question et Xi, Bi, les valeurs de caractères et la longueur transformée des branches ascendantes des noeudsfils du noeud k.
X k=
∑X i
Bi
∑1Bi
Bi =bi1
1b j
1bk
Avec bi, la longueur non transformée de la branche en question et bj, bk les longueurs des branchesfilles.
2. Calcul de l'amplitude de divergencesSoit Xk, la valeur de caractère ancestral du noeud en question et Xi, les valeurs de caractères des noeudsfils (en quantité n) du noeud k. L'amplitude de divergence Ai se calcule ainsi :
Ai=∑i=1
n
X i−X k 2
n
Ainsi appliqué au noeud focal de l'exemple :
22
Figure 9: Exemple d'arbre phylogénétique avec les valeurs de traits des feuilles et l'estimation des caractères ancestraux (en italique)
Noeud k
4 3 2 4 5 2 1 6 4 8 9
3.25 52.67
2.97
3.73
4.82
7
Sousclade 1 Sousclade 2
Clade associé au noeud k

Ai= 2 .97−3.73 2 5−3 .73
2
2=1.05
3.3 Calcul de la contributionCalcul de la quantité de variation au sein du clade associé au noeud k, due à la première divergenceCalcul de la variation due à la divergence du noeud k :
V i=∑i=1
n
N i× X i−X k 2
Application à l'exemple pour le noeud k : V k=7×2 .97−3.73 22×5−3 .73
2=7.27
Calcul de la quantité de variation émergeant dans les sousclades (sans prendre en compte leur structure phylogénétique interne)Soit Lj, les valeurs des feuilles descendantes des noeuds des sousclades considérés et mi, le nombre de feuilles descendantes du noeud i.
V subclades=∑i= 1
n
∑j=1
mi
L j−X i 2
Application à l'exemple : V subclades=2×2−2 .97
2 3−2 .97
22×4−2 .97
2 5−2 .97
2 1 −2 .97
26−5
2 4−5
2
V subclades=1 4 . 0 1Calcul de la quantité de variation au sein du clade associé au noeud k, due à la première divergence :
Vark=V k
V k +V subclades
=0 .34
Calcul de la quantité de la variation totale du trait contenue dans le clade du noeud kQuantité de variation SSk au sein du clade associé au noeud k :
SS k=∑i= 1
mk
L i−X k 2
Application à l'exemple :SS k=1−3 .73
22× 2−3 .73
2 3−3 .73
23× 4−3 .73
25−3.73
26−3.73
2=20 .96
Quantité de variation SS au sein de l'ensemble de l'arbre
SS=∑i= 1
mtot
L i−X Root 2 avec mtot, le nombre de feuilles de l'arbre et Xroot la valeur de caractère
ancestral de la racineApplication à l'exemple :SS= 1−4 .82
22× 2−4 .82
23−4 .82
23× 4−4 .82
2 5−4 .82
26−4. 82
28−4 .82
29−4 .82
2
SS= 60 . 29
Calcul de la contribution :
Contributionk =Var k×SSk
SS=0 .118
23
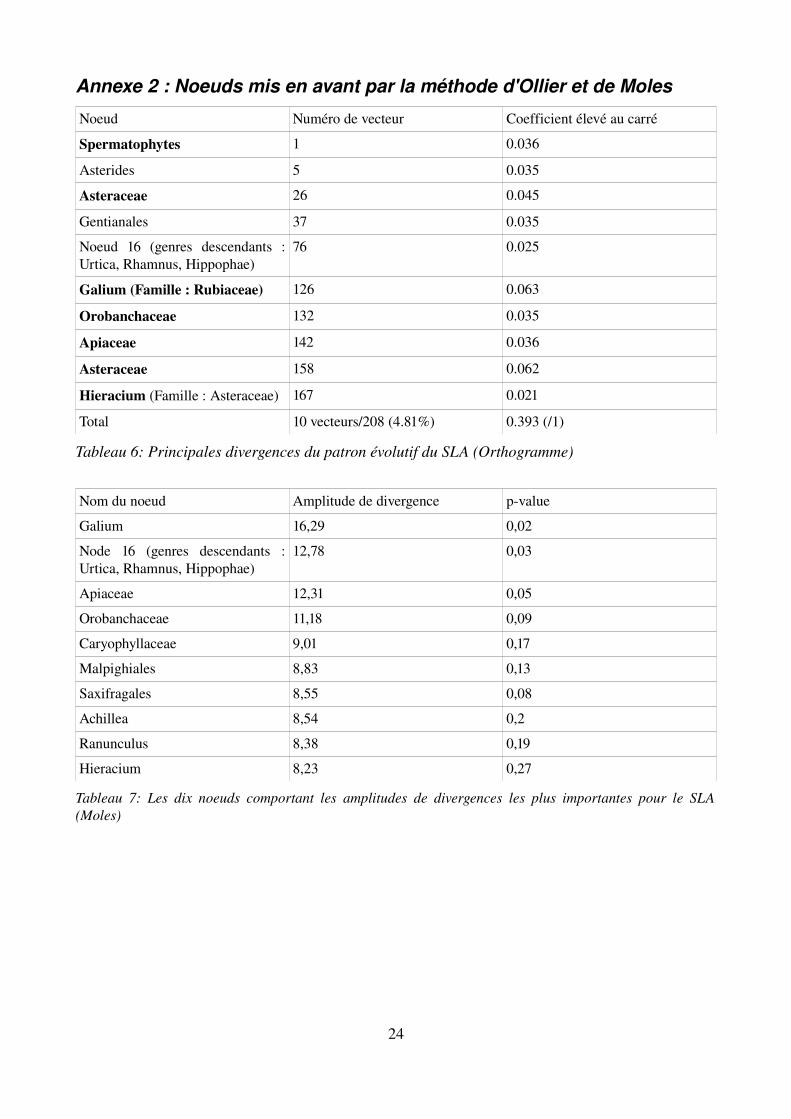
Annexe 2 : Noeuds mis en avant par la méthode d'Ollier et de Moles
Noeud Numéro de vecteur Coefficient élevé au carré
Spermatophytes 1 0.036
Asterides 5 0.035
Asteraceae 26 0.045
Gentianales 37 0.035
Noeud 16 (genres descendants : Urtica, Rhamnus, Hippophae)
76 0.025
Galium (Famille : Rubiaceae) 126 0.063
Orobanchaceae 132 0.035
Apiaceae 142 0.036
Asteraceae 158 0.062
Hieracium (Famille : Asteraceae) 167 0.021
Total 10 vecteurs/208 (4.81%) 0.393 (/1)
Tableau 6: Principales divergences du patron évolutif du SLA (Orthogramme)
Nom du noeud Amplitude de divergence pvalue
Galium 16,29 0,02
Node 16 (genres descendants : Urtica, Rhamnus, Hippophae)
12,78 0,03
Apiaceae 12,31 0,05
Orobanchaceae 11,18 0,09
Caryophyllaceae 9,01 0,17
Malpighiales 8,83 0,13
Saxifragales 8,55 0,08
Achillea 8,54 0,2
Ranunculus 8,38 0,19
Hieracium 8,23 0,27
Tableau 7: Les dix noeuds comportant les amplitudes de divergences les plus importantes pour le SLA (Moles)
24
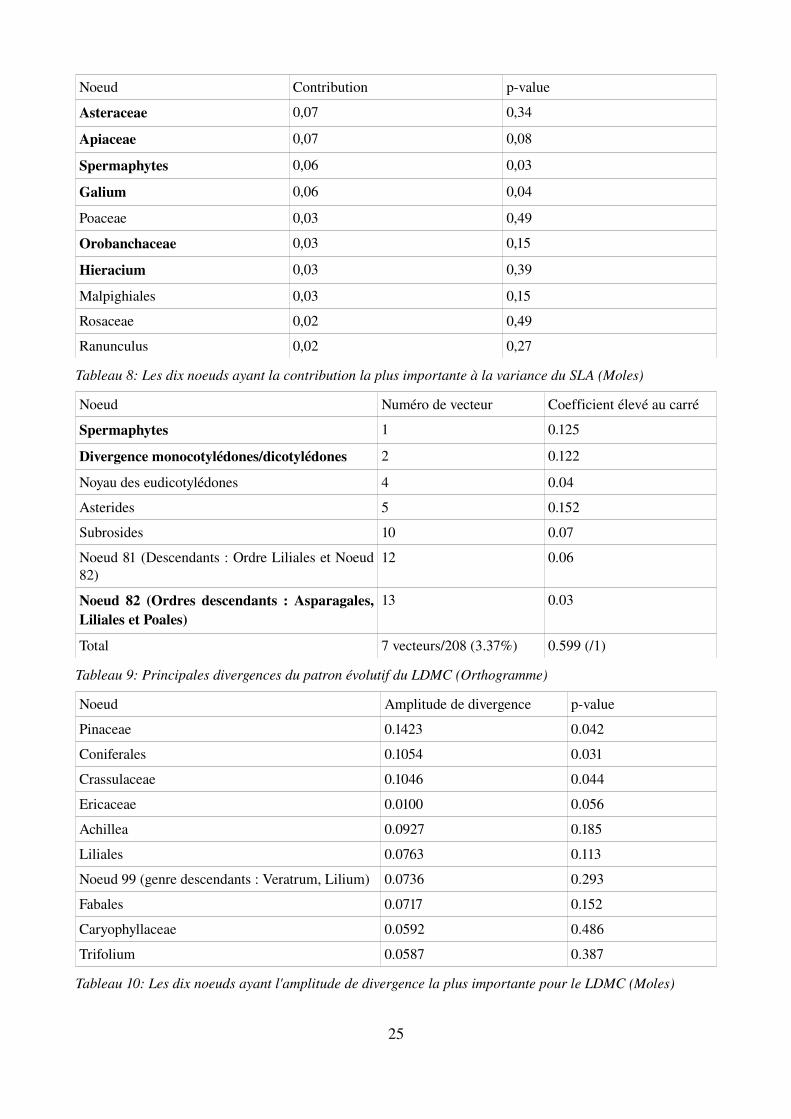
Noeud Contribution pvalue
Asteraceae 0,07 0,34
Apiaceae 0,07 0,08
Spermaphytes 0,06 0,03
Galium 0,06 0,04
Poaceae 0,03 0,49
Orobanchaceae 0,03 0,15
Hieracium 0,03 0,39
Malpighiales 0,03 0,15
Rosaceae 0,02 0,49
Ranunculus 0,02 0,27
Tableau 8: Les dix noeuds ayant la contribution la plus importante à la variance du SLA (Moles)
Noeud Numéro de vecteur Coefficient élevé au carré
Spermaphytes 1 0.125
Divergence monocotylédones/dicotylédones 2 0.122
Noyau des eudicotylédones 4 0.04
Asterides 5 0.152
Subrosides 10 0.07
Noeud 81 (Descendants : Ordre Liliales et Noeud 82)
12 0.06
Noeud 82 (Ordres descendants : Asparagales, Liliales et Poales)
13 0.03
Total 7 vecteurs/208 (3.37%) 0.599 (/1)
Tableau 9: Principales divergences du patron évolutif du LDMC (Orthogramme)
Noeud Amplitude de divergence pvalue
Pinaceae 0.1423 0.042
Coniferales 0.1054 0.031
Crassulaceae 0.1046 0.044
Ericaceae 0.0100 0.056
Achillea 0.0927 0.185
Liliales 0.0763 0.113
Noeud 99 (genre descendants : Veratrum, Lilium) 0.0736 0.293
Fabales 0.0717 0.152
Caryophyllaceae 0.0592 0.486
Trifolium 0.0587 0.387
Tableau 10: Les dix noeuds ayant l'amplitude de divergence la plus importante pour le LDMC (Moles)
25

Noeud Contribution Pvalue
Spermaphytes 0.2486 0.001
Divergence mono/dicotylédones 0.0222 0.006
Crassulacées 0.0209 0.045
Noeud 82 (à l'origine des Poales et des Asperagales/Liliales)
0.0191 0.002
Pinaceae 0.0181 0.099
Ericaceae 0.0174 0.148
Eudicotyledones 0.0148 0.101
Asteraceae 0.0134 0
Malpighiales 0.0107 0.42
Asterales 0.0104 0.116
Tableau 11: Les 10 noeuds ayant la plus grande contribution pour le LDMC
Annexe 3 : Scripts
Ces scripts ont été conçus pour un usage via le logiciel R, les librairies nécessaires à leur bon fonctionnement sont indiqués pour chacun des scripts.
Démarche de Pagelpagel=function(tree,tree.data){
# Nécessite la librairie "geiger" et les libraires associées
## Modèle lambda : tester le signal phylogénétique#lambdaFit < fitContinuous(tree, tree.data, model="lambda")lambdaFit_1 <fitContinuous(tree,tree.data,model="lambda",bounds=list(lambda=c(0.9999,1.0001)))lambdaFit_0 <fitContinuous(tree,tree.data,model="lambda",bounds=list(lambda=c(0.0001,0.0001)))
#Calcul des vraisemblancesLikR.lambda_0<2*(lambdaFit[[1]]$lnllambdaFit_0[[1]]$lnl)Proba_lambda_0<1pchisq(LikR.lambda_0,1,lower.tail="FALSE")
LikR.lambda_1<2*(lambdaFit[[1]]$lnllambdaFit_1[[1]]$lnl)Proba_lambda_1<1pchisq(LikR.lambda_1,1,lower.tail="FALSE")
#Notation des valeurs de lambdalambda<lambdaFit[[1]]$lambdalambda_1<lambdaFit_1[[1]]$lambdalambda_0<lambdaFit_0[[1]]$lambda
#Notation des AICcAICclambda<lambdaFit[[1]]$aiccAICclambda_1<lambdaFit_1[[1]]$aiccAICclambda_0<lambdaFit_0[[1]]$aicc
26

#Tableau finalmodel.lambda=data.frame(matrix(c(lambda,lambda_0,lambda_1,lambdaFit[[1]]$lnl,lambdaFit_0[[1]]$lnl,lambdaFit_1[[1]]$lnl,AICclambda,AICclambda_0,AICclambda_1,1,Proba_lambda_0,Proba_lambda_1),nrow=3,ncol=4),row.names=c("model","Brownian model","Total phylo"))colnames(model.lambda)=c("lambda","LikR","AICc","pvalue")
print("Lambda done")print(model.lambda)
## Modèle kappa : tester le mode phylogénétique#kappaFit < fitContinuous(tree, tree.data, model="kappa")kappaFit_1 <fitContinuous(tree,tree.data,model="kappa",bounds=list(kappa=c(0.9999,1.0001)))kappaFit_0 <fitContinuous(tree,tree.data,model="kappa",bounds=list(kappa=c(0.0001,0.0001)))
#Calcul des vraisemblancesLikR.kappa_0<2*(kappaFit[[1]]$lnlkappaFit_0[[1]]$lnl)Proba_kappa_0<1pchisq(LikR.kappa_0,1,lower.tail="FALSE")
LikR.kappa_1<2*(kappaFit[[1]]$lnlkappaFit_1[[1]]$lnl)Proba_kappa_1<1pchisq(LikR.kappa_1,1,lower.tail="FALSE")
#Notation des valeurs de kappakappa<kappaFit[[1]]$lambdakappa_1<kappaFit_1[[1]]$lambdakappa_0<kappaFit_0[[1]]$lambda
#Notation des AICcAICckappa<kappaFit[[1]]$aiccAICckappa_1<kappaFit_1[[1]]$aiccAICckappa_0<kappaFit_0[[1]]$aicc
#Tableau finalmodel.kappa=data.frame(matrix(c(kappa,kappa_0,kappa_1,kappaFit[[1]]$lnl,kappaFit_0[[1]]$lnl,kappaFit_1[[1]]$lnl,AICckappa,AICckappa_0,AICckappa_1,1,Proba_kappa_0,Proba_kappa_1),nrow=3,ncol=4),row.names=c("model","Puncevol","Brow_model"))colnames(model.kappa)=c("kappa","LikR","AICc","pvalue")
print("Kappa done")
## Modèle delta : tester le tempo phylogénétique#deltaFit < fitContinuous(tree, tree.data, model="delta")deltaFit_1 <fitContinuous(tree,tree.data,model="delta",bounds=list(delta=c(0.9999,1.0001)))deltaFit_0 <fitContinuous(tree,tree.data,model="delta",bounds=list(delta=c(0.0001,0.0001)))
#Calcul des vraisemblancesLikR.delta_0<2*(deltaFit[[1]]$lnldeltaFit_0[[1]]$lnl)Proba_delta_0<1pchisq(LikR.delta_0,1,lower.tail="FALSE")
LikR.delta_1<2*(deltaFit[[1]]$lnldeltaFit_1[[1]]$lnl)Proba_delta_1<1pchisq(LikR.delta_1,1,lower.tail="FALSE")
#Notation des valeurs de delta
27

delta<deltaFit[[1]]$deltadelta_1<deltaFit_1[[1]]$deltadelta_0<deltaFit_0[[1]]$delta
#Notation des AICcAICcdelta<deltaFit[[1]]$aiccAICcdelta_1<deltaFit_1[[1]]$aiccAICcdelta_0<deltaFit_0[[1]]$aicc
#Tableau finalmodel.delta=data.frame(matrix(c(delta,delta_0,delta_1,deltaFit[[1]]$lnl,deltaFit_0[[1]]$lnl,deltaFit_1[[1]]$lnl,AICcdelta,AICcdelta_0,AICcdelta_1,1,Proba_delta_0,Proba_delta_1),nrow=3,ncol=4),row.names=c("model","Punc_evol","Brow_model"))colnames(model.delta)=c("delta","LikR","AICc","pvalue")
print("Delta done")## Résultats#list(model.lambda,model.kappa,model.delta)}
Démarche de Molesmole.node=function(arbre,data,nrepet){#Présupposés : data est un vecteur avec les valeurs de traits avec les noms d'espèces associé, arbre est de type phylo et le label de la racine est "Root".#Librairies : "ape","phylobase"
arbre_di=multi2di(arbre)n=length(arbre_di$node.label)for (i in 1:n){
if (arbre_di$node.label[i]==""){arbre_di$node.label[i]=paste("N",as.character(i),sep="")
}}arbre_di$node=arbre_di$node.labelcontrib_index=vector(mode="numeric",length=length(arbre$node))names(contrib_index)=arbre$nodecorBrow=corBrownian(phy=arbre_di)#Estimate internal node valuesdata.node=ace(data[arbre_di$tip],arbre_di,type="continuous",method="GLS",corStruct=corBrow)$acenames(data.node)=arbre_di$nodetab=c(data.node[arbre$node],data) #Notation des valeurs ancestrales des noeuds de l'arbre originalnames(tab)=c(arbre$node,names(data))arbre.phylo=as(arbre,"phylo4")n=length(arbre$node.label)#Calcul de la contribution de chaque noeudfor (i in 1:n){
contrib_index[arbre$node.label[i]]<moles(arbre.phylo,arbre$node.label[i],tab)indic=paste("Node",as.character(i))print(indic)
}standard_div=vector(length=n)names(standard_div)=arbre$nodep_values_div=standard_divp_values_cont=standard_divcomp_div=standard_div
28

comp_cont=standard_div
#Divergence widthn=length(arbre$node)for (i in 1:n){
u=names(children(arbre.phylo,arbre$node[i]))w=tab[u]tab[arbre$node[i]]standard_div[arbre$node[i]]<(sqrt(sum(w^2)/length(u)))
}p_value_250=data.frame(matrix(,nrow=250,ncol=n))colnames(p_value_250)=arbre$nodep_value_250_cont=p_value_250p_value_500=data.frame(matrix(,nrow=250,ncol=n))colnames(p_value_500)=arbre$nodep_value_500_cont=p_value_500p_value_750=data.frame(matrix(,nrow=250,ncol=n))colnames(p_value_750)=arbre$nodep_value_750_cont=p_value_750p_value_999=data.frame(matrix(,nrow=249,ncol=n))colnames(p_value_999)=arbre$nodep_value_999_cont=p_value_999#Significativité des résultats d'amplitude de divergence et de contribution
#Constitution des divergences et contribution des arbres randomisésfor (j in 1:250){
data=sample(data,length(data))names(data)=arbre$tipdata.node=ace(data[arbre_di$tip],arbre_di,type="continuous",method="GLS",corStruct=corBrow)
$acenames(data.node)=arbre_di$nodetab=c(data.node[arbre$node],data)names(tab)=c(arbre$node,names(data))for (i in 1:n){
u=names(children(arbre.phylo,arbre$node[i]))p_value_250[j,arbre$node[i]]=sqrt(sum((tab[u]tab[arbre$node[i]])^2)/length(u))p_value_250_cont[j,arbre$node[i]]=moles(arbre.phylo,arbre$node[i],tab)
}indica=paste("repet",as.character(j))print(indica)
}for (j in 1:250){
data=sample(data,length(data))names(data)=arbre$tip
data.node=ace(data[arbre_di$tip],arbre_di,type="continuous",method="GLS",corStruct=corBrow)$acenames(data.node)=arbre_di$nodetab=c(data.node[arbre$node],data)names(tab)=c(arbre$node,names(data))for (i in 1:n){
u=names(children(arbre.phylo,arbre$node[i]))p_value_500[j,arbre$node[i]]=sqrt(sum((tab[u]tab[arbre$node[i]])^2)/length(u))p_value_500_cont[j,arbre$node[i]]=moles(arbre.phylo,arbre$node[i],tab)
}indica=paste("repet",as.character(j+250))print(indica)
}for (j in 1:250){
data=sample(data,length(data))names(data)=arbre$tip
29

data.node=ace(data[arbre_di$tip],arbre_di,type="continuous",method="GLS",corStruct=corBrow)$acenames(data.node)=arbre_di$nodetab=c(data.node[arbre$node],data)names(tab)=c(arbre$node,names(data))for (i in 1:n){
u=names(children(arbre.phylo,arbre$node[i]))p_value_750[j,arbre$node[i]]=sqrt(sum((tab[u]tab[arbre$node[i]])^2)/length(u))p_value_750_cont[j,arbre$node[i]]=moles(arbre.phylo,arbre$node[i],tab)
}indica=paste("repet",as.character(j+500))print(indica)}
for (j in 1:249){data=sample(data,length(data))names(data)=arbre$tip
data.node=ace(data[arbre_di$tip],arbre_di,type="continuous",method="GLS",corStruct=corBrow)$acenames(data.node)=arbre_di$nodetab=c(data.node[arbre$node],data)names(tab)=c(arbre$node,names(data))for (i in 1:n){
u=names(children(arbre.phylo,arbre$node[i]))p_value_999[j,arbre$node[i]]=sqrt(sum((tab[u]tab[arbre$node[i]])^2)/length(u))p_value_999_cont[j,arbre$node[i]]=moles(arbre.phylo,arbre$node[i],tab)
}indica=paste("repet",as.character(j+750))print(indica)}
p_value=rbind(p_value_250,p_value_500,p_value_750,p_value_999)p_value_cont=rbind(p_value_250_cont,p_value_500_cont,p_value_750_cont,p_value_999_cont)
#Calcul des pvalue et écart à la randomisationfor (i in 1:n){
v=as.vector(p_value[,arbre$node[i]],mode="numeric")u=split(v,v<standard_div[arbre$node[i]])$"TRUE"p=length(u)/(nrepet+1)if (p>0.5){
p=1p}p_values_div[arbre$node[i]]=pprint(paste("pvalue",as.character(i)))
comp_div[arbre$node[i]]=standard_div[arbre$node[i]]mean(v)
v=as.vector(p_value_cont[,arbre$node[i]],mode="numeric")u=split(v,v<contrib_index[arbre$node[i]])$"TRUE"p=length(u)/(nrepet+1)if (p>0.5){
p=1p}p_values_cont[arbre$node[i]]=p
comp_cont[arbre$node[i]]=contrib_index[arbre$node[i]]mean(v)}time=branching.times(arbre)index=data.frame(standard_div,p_values_div,contrib_index,p_values_cont)print(index)print(comp_div)print(comp_cont)layout(matrix(c(1:2),2,1))plot(x=time[arbre$node],y=comp_div[arbre$node],type="p",xlab="âge du noeud",ylab="D
Drand",main="Amplitude de divergence des noeuds")plot(x=time[arbre$node],y=comp_cont[arbre$node],type="p",xlab="âge du noeud",ylab="C
30

Crand",main="Contribution des noeuds")
list(index,comp_div,comp_cont)}
moles=function(arbre.phylo,node,tab){#Cette fonction permet de calculer pour un noeud donné, sa contribution. Elle est intrinsèque à la fonction mole.node.#Présupposés : node est le nom du noeud visé (mode character), arbre est de type phylo et le label de la racine est "Root". tab est obtenu avec la partie "index" de la fonction totale. C'est un vecteur présentant les valeurs de caractères de chaque noeud et feuille.#librairies nécessaires : "ape","phylobase"
#Calculate the amount of variation within the focal clade that is attributable to the focal divergence#variation within the subclades
#focal divergenceu=children(arbre.phylo,node)n=length(u)var.focal=0var.clades=0a=tab[node]names(a)=c()for (i in 1:n){
y=tab[names(u)[i]]names(y)=c()desc=length(descendants(arbre.phylo,u[i]))var.focal=var.focal+desc*(ya)*(ya);w=descendants(arbre.phylo,u[i])if (mode(w)!="character"){#c'estàdire que u[i] est un noeud et pas une feuille.
m=length(w)z=tab[names(u)[i]]names(z)=c()for (j in 1:m){
b=tab[names(w)[j]]names(b)=c()var.clades=var.clades+(bz)*(bz);}}}
var.clade.first.divergence=var.focal/(var.focal+var.clades)#Calculate the amount of the total variation across the whole tree that lies within the focal clade
u2=descendants(arbre.phylo,as.numeric(getnodes(arbre.phylo,node)))n=length(u2)SS=0for (i in 1:n){
x=tab[names(u2)[i]]names(x)=c()SS=SS+(xa)*(xa);
}u3=arbre.phylo$tip.labelb=tab["Root"]names(b)=c()n=length(u3)SS_tot=0for (i in 1:n){
x=tab[u3[i]]SS_tot=SS_tot+(xb)*(xb);}
prop_var=SS/SS_tot#Calculate the contribution index
focal_divergence=var.clade.first.divergence*prop_varfocal_divergence}
31


















