
chimiomtrie2
18
Chimiométrie 1. INTRODUCTION La chimiométrie est un outil utilisé afin d’extraire de l’information pertinente et utile à partir de données physico- chimiques mesurées ou connues brutes. Il est basé sur la construction, puis l’exploitation d’un modèle de comportement à l’aide d’outils statistiques. Il peut traiter des systèmes complexes et donc généralement multivariables. Le terme « chimiométrie » vient de l’anglais « chemometrics », jeune discipline associant initialement analyse de données et chimie analytique. Aujourd’hui, il recouvre l’ensemble des applications de la chimie, de la physique, des sciences de la vie, de l’économie, de la sociologie, des méthodes statistiques et de l’informatique. Aussi, on lui préfère souvent le terme moins restrictif (pa r rapport à la chimie) «d’analyse multivariable » (« multi vari ate ana lysi s »), voire de reconnaissance de forme au sens large (« pattern recognition »). Nous nous intéres serons plus particu lièremen t au x ap plicati ons « origine lles » de la chimiométrie dans le domaine de la chimie analytique, et plus particulièrement des multicapteurs ou capteurs multivariables. La chimiométrie (ou analyse multivariable) en instrumentation consiste à modéliser les variations d’un certain nombre de variables, que nous appellerons Yvariables dont l’obtention est délicate (nécessitant une analyse chimique par exemple) en fonction d’autres variables appelée s Xvariables mesurable s «facileme nt » (mesure de capteu rs physiques par exemp le) afin de pouvoir se passer ultérieu rement de l’obtenti on des premièr es. On distingue 2 opérations: - L 'étalonnage en laboratoire (ou modélisation) où toutes les mesures de variables doivent être réalisées et où le modèle (ou « prédicteur ») est calculé. Attention, ce terme se traduit en anglais par « calib ration », à ne pas confondre avec le « calib rage » d’un instrument qui est le recalage à l’aide d’un ou deux mesurages (« gauging » en anglais). - La prédiction, utilisation cour ante « sur le te rrai n », où seules les variables X « fac ile s » sont mesurées , les autres, Y, étant calculées à l’aide du modèle. ET ALONNAGE / MODELISATION PREDICTION Variables X mesurées (capteurs) Variables Y mesurées ana l ses Calcul de modélisation Prédicteur Variables X mesurées (capteurs) Prédicteur Calcul de prédiction Estimation des variables Y 1.1. Pr oblè me de l’ anal ys e mult ivar iabl e: Soient m échantillons , dits d'étalonnage, chacun , chacun étant cara ctérisé par c Yvariables caractéristique s, par définition no n « facilement » mesurables directement (par exemple des concentrations chimiques), mais dont une évaluation y j est supposée connue pendant la phase d'étalonnage: c j m i y Y i j ≤ ≤ ≤ ≤ = 1 1 , Par contre, n Xvariables sont « facilement » mesurables (par exemple des tensions), leur mesure est appelée x k : ( ) n k m i x X i k ≤ ≤ ≤ ≤ = 1 1 , Le problème consiste, à partir de ces échantillons d'étalonnage, à trouver un modèle, c’est à dire une fonction multivariables F telle que pour tout autre échantillon on puisse évaluer, à partir des seules Xvariables X :
-
Upload
samantha-farah -
Category
Documents
-
view
218 -
download
0
Transcript of chimiomtrie2

7/22/2019 chimiomtrie2
http://slidepdf.com/reader/full/chimiomtrie2 1/18
Chimiométrie
1. INTRODUCTION
La chimiométrie est un outil utilisé afin d’extraire de l’information pertinente et utile à partir de données physico-chimiques mesurées ou connues brutes. Il est basé sur la construction, puis l’exploitation d’un modèle de comportement
à l’aide d’outils statistiques. Il peut traiter des systèmes complexes et donc généralement multivariables.
Le terme « chimiométrie » vient de l’anglais « chemometrics », jeune discipline associant initialement analyse de
données et chimie analytique. Aujourd’hui, il recouvre l’ensemble des applications de la chimie, de la physique, des
sciences de la vie, de l’économie, de la sociologie, des méthodes statistiques et de l’informatique.
Aussi, on lui préfère souvent le terme moins restrictif (par rapport à la chimie) «d’analyse multivariable »
(« multivariate analysis »), voire de reconnaissance de forme au sens large (« pattern recognition »).
Nous nous intéresserons plus particulièrement aux applications « originelles » de la chimiométrie dans le domaine
de la chimie analytique, et plus particulièrement des multicapteurs ou capteurs multivariables.
La chimiométrie (ou analyse multivariable) en instrumentation consiste à modéliser les variations d’un certain
nombre de variables, que nous appellerons Yvariables dont l’obtention est délicate (nécessitant une analyse chimique
par exemple) en fonction d’autres variables appelées Xvariables mesurables «facilement » (mesure de capteurs
physiques par exemple) afin de pouvoir se passer ultérieurement de l’obtention des premières.
On distingue 2 opérations:
- L'étalonnage en laboratoire (ou modélisation) où toutes les mesures de variables doivent être réalisées et où le
modèle (ou « prédicteur ») est calculé. Attention, ce terme se traduit en anglais par « calibration », à ne pas confondre
avec le « calibrage » d’un instrument qui est le recalage à l’aide d’un ou deux mesurages (« gauging » en anglais).
- La prédiction, utilisation courante « sur le terrain », où seules les variables X « faciles » sont mesurées , les autres,
Y, étant calculées à l’aide du modèle.
ETALONNAGE / MODELISATION PREDICTION
Variables X
mesurées
(capteurs)
Variables Y
mesurées
anal ses
Calcul de
modélisation
Prédicteur
Variables X
mesurées
(capteurs)
Prédicteur
Calcul de
prédiction
Estimation des
variables Y
1.1. Problème de l’ anal yse mult ivar iabl e:
Soient m échantillons, dits d'étalonnage, chacun , chacun étant caractérisé par c Yvariables caractéristiques, par
définition non « facilement » mesurables directement (par exemple des concentrations chimiques), mais dont une
évaluation y j est supposée connue pendant la phase d'étalonnage:
c jmi yY i
j ≤≤≤≤= 1 1 ,
Par contre, n Xvariables sont « facilement » mesurables (par exemple des tensions), leur mesure est appelée xk :
( ) nk mi x X i
k ≤≤≤≤= 1 1 ,
Le problème consiste, à partir de ces échantillons d'étalonnage, à trouver un modèle, c’est à dire une fonction
multivariables F telle que pour tout autre échantillon on puisse évaluer, à partir des seules Xvariables X :

7/22/2019 chimiomtrie2
http://slidepdf.com/reader/full/chimiomtrie2 2/18
2
Y = F(X) où F est appelé prédicteur.
Deux types de modèle sont envisageables :
- Le « modèle de connaissance », réalisé à partir des connaissances physico-chimiques que l’on a du
problème. Mais cette approche, déjà complexe et approximative (d’un point de vue instrumentation) lorsqu’il
s’agit d’exprimer les Xvariables en fonction des Yvariables, s’avère généralement impraticable dans le cas
inverse (celui qui nous intéresse : Y=F(X)), notamment à cause de la complexité des fonctions, du bruit de
mesure et des effets d’impuretés non mesurés. On lui préfère classiquement :
- Le « modèle de comportement » : On ne s’intéresse qu’au modèle mathématique permettant de reproduire
« au mieux » les relations entre X et Yvariables d’étalonnage. La connaissance physico-chimique du
problème n’est alors plus nécessaire, c’est un modèle du type « boite noire » .
Toutefois il est généralement préférable dans le cas du modèle de comportement qu’un minimum de connaissance
théorique soit disponible (du genre « le problème est-il linéaire ? ») Mais nous verrons à la fin du chapitre que les réseaux
neuronaux permettent de se passer complètement du modèle de connaissance.
1.2. Exemples:
Exemple 1 (trivial): Mesure de la caractéristique d'une pile électrique:
On mesure quelques couples de points (U, I), or on sait que ces points doivent suivre une loi du type:
U = E -rI où E est la f.e.m. de la pile et r sa résistance interne (partie « modèle de connaissance »).
r
pile
U
I
I
U
E
On peut alors faire une estimation des valeurs de E et de r par "régression linéaire", en tenant compte de tous les
couples (U, I) pour une meilleure précision. On a alors une connaissance quantitative du phénomène. De plus, on
peut par la suite calculer la valeur de U à partie de la seule mesure de I (phase prédiction).
Il s'agit ici "d'analyse mono variable".
Exemple 2: Loi d'absorption de la lumière dite de Beer-Lambert :
Source
polychromatique
Echantillon
(Solution)
Spectrophotomètre
La spectrophotométrie cocnsiste à mesurer à travers une solution contenant un composé dissous absorbant la
lumière, "l'atténuation" de l'intensité d'une source lumineuse: on appelle absorbance à la longueur λk la grandeur:
log ref
10
=
k
k
k I
I A où I k ref est l’intensité de référence.

7/22/2019 chimiomtrie2
http://slidepdf.com/reader/full/chimiomtrie2 3/18
3
La loi de Beer Lambert nous indique que cette grandeur est proportionnelle à la concentration du composé:
A m C k k =
Si on mélange plusieurs composés, les absorbances sont additives:
Pour tous les λk k k
j
j
ja m c, = ∑
L'analyse multivariable permettra, après étalonnage avec des échantillons de concentrations connues, de
trouver les concentrations d'autres échantillons à partir des seules mesures d'absorbances.
C’est probablement l’utilisation la plus classique de la chimiométrie, aussi, tous les exemples l’utiliseront, la
matrice A des absorbances sera celle des Xvariables et la matrice C des concentrations celle des Yvariables.
Poi nt commun àtou tes les méthodes:
* On suppose que le modèle qui décrit les échantillons d’étalonnage, décrit aussi le phénomène physico-chimique
que l'on veut mesurer et est donc applicable pour la phase de prédiction.
* Attention: un "bon" modèle pour les échantillons d’étalonnage n'en est pas forcément un pour d'autres
échantillons (de prédiction). En effet, il est toujours possible, en augmentant suffisamment la complexité du modèle, de
faire en sorte que celui-ci reproduise aussi bien qu'on le voudra les variations des jeux de variables d’étalonnage. On
aura alors modélisé non seulement les phénomènes physiques, mais aussi les bruits de mesure et autres sources
d'incertitude. Ce problème, dit de sur-modélisation, sera développé plus loin.
* Il est donc difficile de définir le critère "au mieux". Seules les méthodes complexes, notamment d'analyse de
facteurs, permettent de faire cette modélisation "efficace" (c'est à dire ne modélisant, dans la mesure du possible que les
phénomènes physiques utiles).
1.3. Méthode générale:
On désire trouver Y=F(X) le "plus précisément possible". On peut donc écrire, pour les échantillons d'étalonnage:
Y=F(X)+E
où E est la matrice d'erreur sur les variables Y que l'on désire minimiser:
( ) E Y F X y f x x j
i
j
i
j
i= − = −( ) ( ,..., )1 minimale (?)
Le concept de minimisation d'une matrice nécessite d'introduire une distance. On utilise généralement la distance
Euclidienne:
( ) y f x x j
i
j
i
k
i
i j
−∑ ( ,..., ),
1
2
minimale
(D'où le nom de méthode des "moindres carrés"; en fait, d'autres types de distances pourraient faire l'affaire, mais
alors le cas linéaire n'aurait pas de solution analytique simple)
Supposons, pour simplifier la formulation qu'il n'y ait qu'une variable y.
La forme générale des fonctions f est supposée connue. On doit donc trouver les constantes a 1,...,aq,...,a p qui les
caractérisent quantitativement, et donc telles que:
( ) y f a a x xi
p
i
k
i
i
−∑ ( ,..., , ,... , )1 1
2
minimale
Donc, pour tout q,
( )
1
1 1
2
≤ ≤
−
∑
q p
y f a a x x
a
i
p
i
k
i
i
q,
( ,..., , ,..., )
∂
∂ = 0
On obtient donc p équations à p inconnues dont la résolution littérale est impossible dans le cas général.

7/22/2019 chimiomtrie2
http://slidepdf.com/reader/full/chimiomtrie2 4/18
4
2. Cas particulier: cas linéaire
2.1. Notati ons & prélim in ai res
Les relations entre les X et les Y sont linéaires et les a p forment une donc matrice A:
Y=XA + E
soit: y x a e j
i
k
i
j
k
j
i
k
= +∑
avec:
1
1
1
≤ ≤≤ ≤≤ ≤
i m
k n
j c
, chantillon,
, num ro de Xvariable,
, num ro de Yvariable
numero d'e
e
e .
(On pourra, si nécessaire, lire les rappels d'algèbre linéaire en annexe).
Remarque: La convention chimiométrique pour l'écriture des matrices, sera utilisée: Chaque ligne représentera un
échantillon (ou expérience) et chaque colonne, une variable.
Remarque 1: cas où Y non nulle pour X=matrice nulle (terme d'offset):
y a a xi
k k
i
k
= + ∑0
Ce cas peut se résoudre directement par la méthode des moindres carrés, mais on préfère
généralement prendre les variables dites centrées:
étalonnaged' x x x
y y y
moyennesvaleurslesétantxety ,*
*
−=
−=
On a alors: y a xi
k k
i
k
* *= ∑
Remarque 2
Les variables y1...y j, ou x1…xk , ne sont pas forcément du même ordre de grandeur, ni même
homogènes: on doit donc parfois les normer avant le étalonnage, c'est à dire les diviser par la racine
carrée de la moyenne de leur carré (norme):
∑=
i
i j
i
ji
j
yn
y y
2)(1
ˆ
Il est donc souvent (mais pas toujours…) préférable de normer puis centrer toutes les variables (A et C) avant
étalonnage. Après la prédiction, les variables C calculées devront donc être décentrées et dénormées avec les valeurs de
moyenne et de norme obtenues lors du étalonnage.
Dans d’autres cas, afin de donner moins d’influence aux variables « bruitées », on norme en divisant par l’écart
type des variables.
Remarque 3 : Matrice de covariance
La matrice X'X (notée aussi X
T
X)que l'on est amené à inverser s'écrit , si les variables x
i
k sontcentrées:

7/22/2019 chimiomtrie2
http://slidepdf.com/reader/full/chimiomtrie2 5/18
5
X X
x x
x x
x x
x x
x x x
x x x
n
x Cov x x
Cov x x x
T
m
n n
m
n
m
n
m
i
i
n
i i
i
i
n
i
i
n
i
i
n
n n
=
=
= −
∑ ∑
∑ ∑
1
1
1
1
1
1 1
1
1
2
1
1
2
2
1 1
1
2
1
L
M O M
L
L
M O M
L
L
M O M
L
L
M O M
L
( )
( )
( )
( ) ( , )
( , ) ( )
σ
σ
Cette matrice symétrique, qui comporte les variances des Xvariables (ou Yvariables) sur la diagonale et les
covariances ailleurs, est appelée matrice des covariances. Elles importante car elle contient des information importantes
et surtout les méthodes de régressions imposent d’inverser soit X’X soit Y’Y
Si 2 colonnes de X sont colinéaires, c'est à dire si pour tous les échantillons 2 variables xk ont des valeurs
proportionnelles, la matrice de covariance ne sera pas inversible.
Application : Exemple de la loi de Beer Lambert
Reprenons l'exemple de la colorimétrie et de la loi de Beer Lambert. On appelle:
aik = absorbance de l'échantillon i à la longueur d'onde λk
ci j = concentration de l'échantillon i en composé j
( ) ( ) ( )1 1 1≤ ≤ ≤ ≤ ≤ ≤k n j c i m, , ,
La loi de Beer Lambert s'écrit, sous forme matricielle:
La loi de Beer Lambert servira de support pour la suite de l'exposé, mais les applications de l'analyse multivariables
ne se limitent pas à la colorimétrie et sont innombrables.
2.2. Méthodes de moindr es car rés simples:
2.2.1. Première méthode: ILS (Inverse Least Square) ou Pmatrice:
La loi de Beer-Lambert est linéaire, on peut l’écrire de façon inversée (d’où le nom de la méthode, la méthode dites
« classique», plus complexe, est expliquée plus loin) en exprimant les concentrations en fonction des absorbances:
C = AP + E
Afin d’illustrer la méthode, il est intéressant de faire une représentation dans l’espace dit « des échantillons » :
Supposons que l'on ait 3 échantillons d'étalonnage caractérisés par 2 Xvariables absorbances A1, A2 et une
Yvariable concentration C. On peut représenter chacune de ces variables par un vecteur dans "l'espace des échantillons"
où chaque dimension du repère représente un échantillon, on a donc ici un espace de dimension 3 représenté en
perspective. Les 3 composantes de chaque vecteur (A ou C) représentent les valeurs de la variable pour les 3
échantillons. Ainsi, 2 variables indépendantes auront pour représentation 2 vecteurs orthogonaux:
j
k
i
j
i
k K C A .=
Matrice des absorbances Matrice des coefficients de Beer-Lambert
Matrice des concentrations

7/22/2019 chimiomtrie2
http://slidepdf.com/reader/full/chimiomtrie2 6/18
6
A 1
A 2
2211 A A
rrαα +
E
Lorsque l’on fait un régression linéaire (régression de C sur A), on cherche à écrire C sous forme d’une
combinaison linéaire de A1 et A2 (c=α1A1+α2A2) : la chose est à priori impossible puisque C r
n'est pas dans le plan
),( 21 A Arr
, mais on va s'en approcher en minimisant le vecteur E r
, différence entre C r
et la combinaison linéaire
approchée (dans le plan ),( 21 A Arr
). E r
sera minimal quand il sera orthogonal au plan ),( 21 A Arr
, et la « modélisation » de
C r
sera sa projection sur ce même plan.
On remarque alors que faire une régression de C sur A consiste à projeter C r
sur le plan des Ar
. P est alors la
matrice de projection dans le plan des Ar
.
L'étalonnage sera d'autant meilleur que C r
sera près du plan des Ar
. E r
représente les variations de C non corrélées
avec celles de A et donc pas expliquées par le modèle, on l’appelle « résidus d’absorbance ».
On minimise ici l'erreur E sur les concentrations, on régresse donc les concentrations sur les absorbances . On peut
montrer que la matrice P, opérateur projection, s’écrit :
P = (A’A)-1A’C qui est la matrice de prédiction
Avantage majeur de cette méthode :
On peut voir facilement que, pendant l’étalonnage, le calcul des termes de P concernant le composé j (= colonnes
de P) ne dépendent pas des concentrations des autres composés.
On n’est donc pas obligé de connaître les concentrations de tous les composés. Les composés présents mais de
concentration inconnue pendant l’étalonnage peuvent être considérés comme des impuretés.
On dit que méthode résout le problème des impuretés , mais celles-ci doivent être présentes de façon significative
dans les échantillons d’étalonnage.
De façon plus générale, il résout le problème des variations dues à des causes externes, celles-ci n’ont pas à être
quantifiées pendant la phase d’étalonnage, mais doivent, répétons-le, être présentes de façon « significative ».
7/22/2019 chimiomtrie2
http://slidepdf.com/reader/full/chimiomtrie2 7/18
7
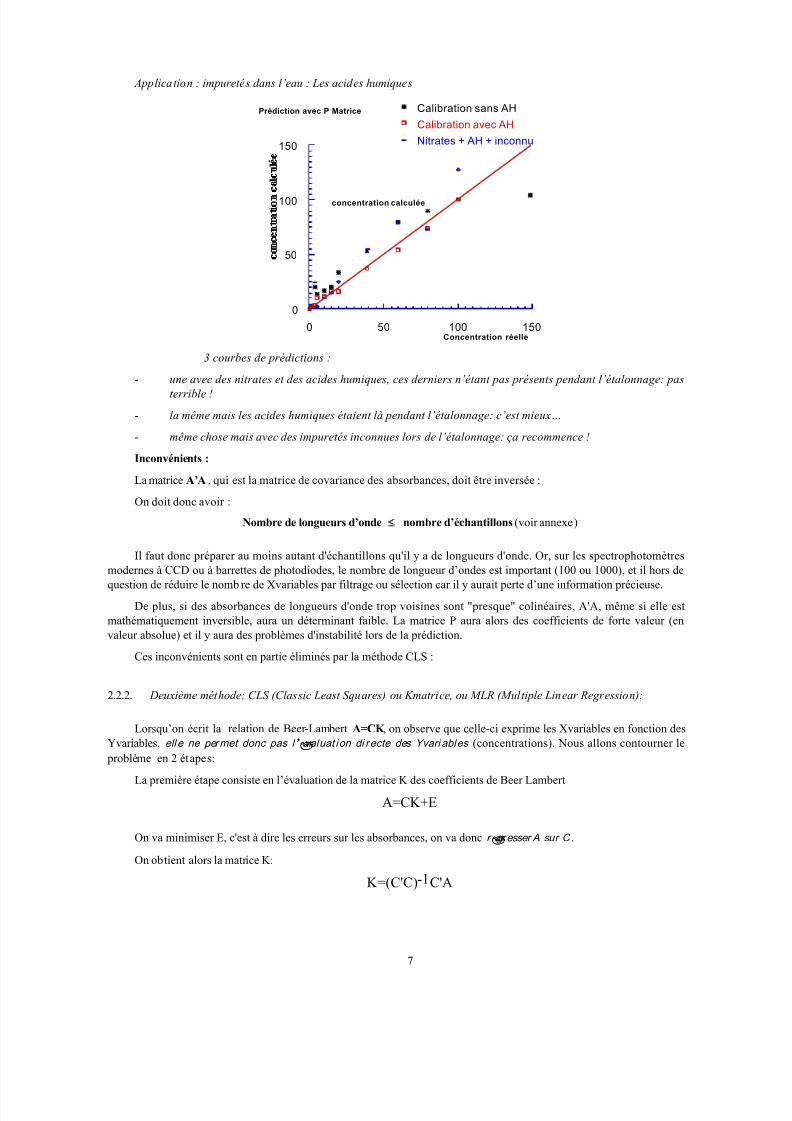
Application : impuretés dans l’eau : Les acides humiques
0
50
100
150
0 50 100 150
concentration calculée
Calibration sans AH
Calibration avec AH
Nitrates + AH + inconnu
Prédiction avec P Matrice
Concentration réelle
3 courbes de prédictions :
- une avec des nitrates et des acides humiques, ces derniers n’étant pas présents pendant l’étalonnage: pas
terrible !
- la même mais les acides humiques étaient là pendant l’étalonnage: c’est mieux…
- même chose mais avec des impuretés inconnues lors de l’étalonnage: ça recommence !
Inconvénients :
La matrice A’A , qui est la matrice de covariance des absorbances, doit être inversée :
On doit donc avoir :
Nombre de longueurs d’onde ≤≤ nombre d’échantillons (voir annexe)
Il faut donc préparer au moins autant d'échantillons qu'il y a de longueurs d'onde. Or, sur les spectrophotomètres
modernes à CCD ou à barrettes de photodiodes, le nombre de longueur d’ondes est important (100 ou 1000), et il hors de
question de réduire le nomb re de Xvariables par filtrage ou sélection car il y aurait perte d’une information précieuse.
De plus, si des absorbances de longueurs d'onde trop voisines sont "presque" colinéaires, A'A, même si elle est
mathématiquement inversible, aura un déterminant faible. La matrice P aura alors des coefficients de forte valeur (en
valeur absolue) et il y aura des problèmes d'instabilité lors de la prédiction.
Ces inconvénients sont en partie éliminés par la méthode CLS :
2.2.2. Deuxième méthode: CLS (Classic Least Squares) ou Kmatrice, ou MLR (Multiple Linear Regression):
Lorsqu’on écrit la relation de Beer-Lambert A=CK , on observe que celle-ci exprime les Xvariables en fonction des
Yvariables, ell e ne permet donc pas l ’évaluat ion di recte des Yvari ables (concentrations). Nous allons contourner le
problème en 2 étapes:
La première étape consiste en l’évaluation de la matrice K des coefficients de Beer Lambert
A=CK+E
On va minimiser E, c'est à dire les erreurs sur les absorbances, on va donc régresser A sur C .
On obtient alors la matrice K:
K=(C'C)-1C'A

7/22/2019 chimiomtrie2
http://slidepdf.com/reader/full/chimiomtrie2 8/18
8
En phase prédiction, nous avons: A=CK , mais c'est la matrice C que l'on veut déterminer.
Or K est généralement non carrée donc non inversible. On peut quand même écrire:
C = AK'(KK')-1 = AM
M est alors la matrice de prédiction.
On est amené à effectuer 2 inversions de matrices carrées de faible dimension (dimension = nombre de composés):
- C'C, matrice de covariance des concentrations : on doit donc avoir:
nombre de composés ≤≤ nombre d'échantillons.
- KK': on doit donc avoir: nombre de composés ≤≤ nombre de longueurs d'onde.
Ces 2 conditions, à priori évidentes, sont faciles à respecter.
Remarque: choix des concentrations:
On remarque que C'C est la matrice de covariance des concentrations. Si 2 colonnes de C sont
colinéaires, C'C ne sera pas inversible, d'où des précautions à prendre pour le choix des concentrations
des échantillons d’étalonnage. De même, si 2 colonnes sont presque colinéaires,(C'C)-1 comprendra de
fortes valeurs et on aura alors des instabilités pendant la prédiction.
Si il n'y a qu'un seul composé, C'C est un scalaire égal à la variance des concentrations et le
problème ne se pose pas.
Avantages de la méthode MLR:
* On peut utiliser théoriquement autant de longueurs d'onde que l'on veut. Le fait d'en avoir beaucoup donne
un effet de "moyenne" bénéfique au niveau rapport signal / bruit.
* Intéressant au niveau compréhension des phénomènes: la matrice K donne directement les coefficients de
Berr Lambert.
Inconvénients de la méthode:* Le calcul des paramètres de prédiction pour un composé utilise les concentrations de tous les composés: En
effet, on est amené à faire une projection orthogonale de chaque absorbance sur l’hyperespace des concentrations. Le
résultat (opérateur projecteur) dépendra donc de la pris e en compte des composés ayant une influence sur les
absorbances. Tous les composés ou interférents voire tous les événements susceptibles d'être présents en phase de
prédiction devront être introduits pendant l’étalonnage, et leur concentration ou valeur numérique devra être connue
sous forme d’un Yvariable.
Cette méthode ne résout donc pas le problème des impuretés.
Pour des raisons similaires, la méthode ne peut pas tenir compte de variations non quantifiées ou non quantifiables
(variation de la ligne de base par exemple), ou d'interactions entre les constituants.
2.2.3. Problème commun aux 2 méthodes : la surmodélisation.
Surmodélisation et erreur de modélisation
La surmodélisation apparaît lorsque le modèle est trop précis, on tend alors à modéliser les "particularités" des
échantillons d'étalonnage, alors que seule l'information "commune" et « utile » est intéressante. On introduit une erreur
de modél isation (différence entre le modèle et la loi réelle, à priori inconnue).
le modèle, trop complexe, ne s’appliquera
correctement qu’aux échantillons ayant servi à
le créer .
Exemple trivial : la régression
polynomiale
Soit une loi physique (courbe grasse) à
modéliser à l’aide de 5 expériences (points
Y
X
loi physique
p=1
sous -modélis atio n p=4
su r-modélis atio n
p=2
OK Y
X
loi physique
p=1
sous -modélis atio n
p=1
sous -modélis atio n
p=1
sous -modélis atio n p=4
su r-modélis atio n
p=4
su r-modélis atio n
p=4
su r-modélis atio n
p=2
OK
7/22/2019 chimiomtrie2
http://slidepdf.com/reader/full/chimiomtrie2 9/18
9
noirs) par régression polynomiale dont on va choisir le degré p:
P=1 : l’erreur de modélisation (différence avec la loi réelle) est forte, le modèle est trop simple, on fait de la
surmodélisation
P=5 : par rapport aux échantillons, c’est excellent, mais l’erreur de modélisation est très importante : on fait de
la surmodélisation.
P=2 : semble être un bon compromis.
Remarque : Si l’on rajoute des points expérimentaux (points gris), alors même un polynôme de degré 5 sera très
proche de la courbe réelle, donc le danger de surmodélisation diminue lorsque le nombre d’expériences (ou
échantillons) d’étalonnage augmente.
Surmodélisation et erreur liée à la mesure :
Lorsque l’on effectue le calcul de prédiction, à partir du modèle et des Xvariables, l’erreur aléatoire de mesure sur
les Xvariables (caractérisée par l’incertitude) se propage à travers le modèle et contribue (avec l’erreur de modélisation) à
l’erreur sur les Yvariables. Cette composante d’erreur est appelée « erreur liée à la mesure ».
Mesure de X : X X ∆+& ( X & = valeur réelle, ∆X = erreur)
Contribution de ∆X à l’erreur sur Y : ∆Yme s
Erreur sur Y : ∆Yme s + ∆Ymod,
Soit, en raisonnant sur les écart-types :22
modY Y Y mesσσσ +=
On peut constater, lors de la prédiction, que même si le modèle est correct physiquement (erreur de modélisation
négligeable) plus un modèle est complexe, plus les erreurs (relatives) liées à la mesure seront amplifiées lors du calcul
des Yvariables :
X
X
Y
Y mes ∆=
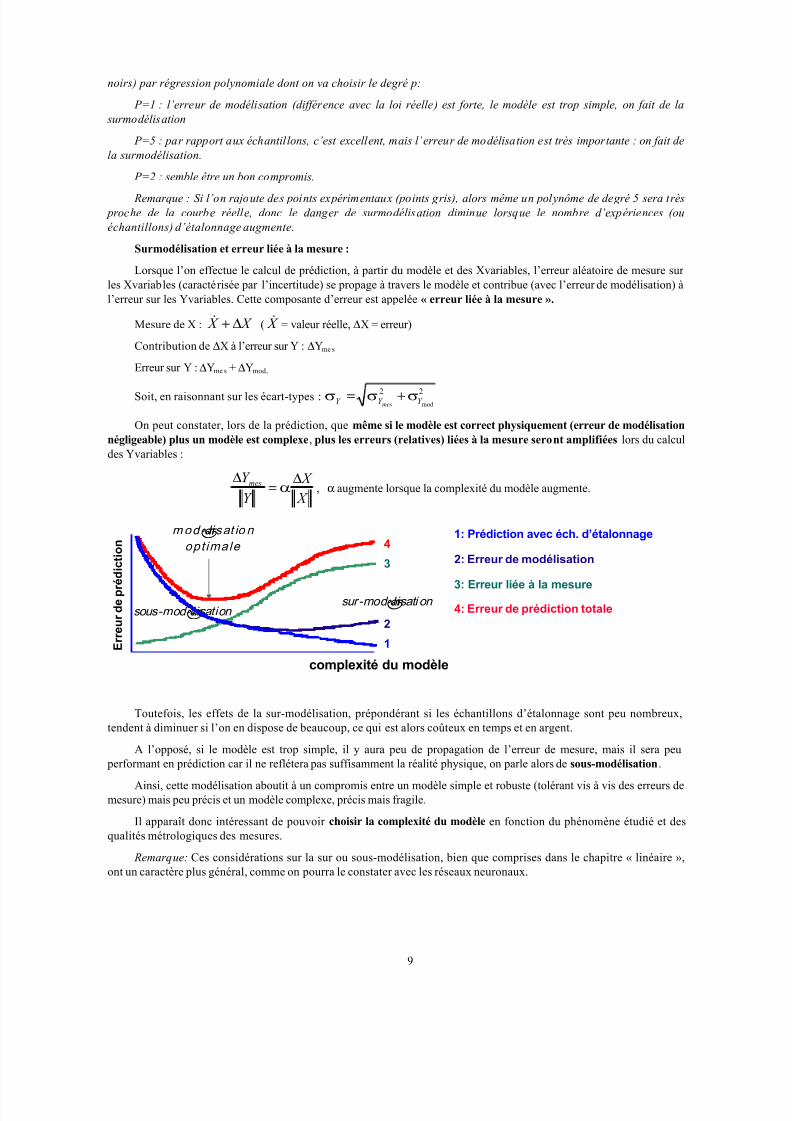
∆α , α augmente lorsque la complexité du modèle augmente.
2: Erreur de modélisation
2
3: Erreur liée à la mesure
3
sur-modélisati on sous-modélisation
modélisat io n opt imale
4: Erreur de prédiction totale
4
complexité du modèle
1: Prédiction avec éch. d’étalonnage
1 E r r e u r d e p r é d i c t i o n
Toutefois, les effets de la sur-modélisation, prépondérant si les échantillons d’étalonnage sont peu nombreux,tendent à diminuer si l’on en dispose de beaucoup, ce qui est alors coûteux en temps et en argent.
A l’opposé, si le modèle est trop simple, il y aura peu de propagation de l’erreur de mesure, mais il sera peu
performant en prédiction car il ne reflétera pas suffisamment la réalité physique, on parle alors de sous-modélisation.
Ainsi, cette modélisation aboutit à un compromis entre un modèle simple et robuste (tolérant vis à vis des erreurs de
mesure) mais peu précis et un modèle complexe, précis mais fragile.
Il apparaît donc intéressant de pouvoir choisir la complexité du modèle en fonction du phénomène étudié et des
qualités métrologiques des mesures.
Remarque: Ces considérations sur la sur ou sous-modélisation, bien que comprises dans le chapitre « linéaire »,
ont un caractère plus général, comme on pourra le constater avec les réseaux neuronaux.
7/22/2019 chimiomtrie2
http://slidepdf.com/reader/full/chimiomtrie2 10/18
10
2.3. B - L 'anal yse de facteur s:
L'analyse de facteurs consiste à faire un changement de variable au niveau des Xvariables. On prend alors, comme
nouvelles Xvariables, des combinaisons linéaires des anciennes, en nombre réduit afin de « concentrer » l'information
utile.
Il y a, initialement, n Xvariables, généralement plus ou moins corrélées entre elles. Le but est de trouver de
nouvelles variables indépendantes indépendantes par changement de base, avec réduction de la dimension. Ces
variables sont appelées facteurs ou variables latentes et leur valeur pour un échantillon donné, score.
A T
dimension n,
corrélées
dimension h n,
ariables
indé endantes
matrice de
changement de
ase B
ariables
Si h=n, il n'y a pas de réduction de données, le problème est équivalent au précédent et la méthode a peu d'intérêt.
Il y a plusieurs méthodes pour déterminer la matrice de changement de base B ("Loadings vecteurs") qui seront
explicitées plus loin.
Pendant la prédiction, le changement de base permettant de connaître les scores T se fait comme suit:
T=B*A (B matrice h*n)
Il est clair que comme h<n, il y aura perte d'information lors du passage de A à T. On va donc faire en sorte que
cette information perdue soit le moins possible de l'information utile:
n n
info
bruit
bruitinfo
h
On peut ensuite calculer les concentrations par régression de T à partir d'une matrice de prédiction V déterminée,
pendant l’étalonnage, par ILS à partir des scores:
C = TV + E
On fait alors une régression de C sur V.
L'intérêt de cette compression de données est que, faute de pouvoir éliminer le bruit de mesure, on s'arrange pour
que l'information utile (qui pouvait être initialement répartie sur tout le spectre) soit dans les premiers facteurs qui seront
Répartition homogène "bruit" de mesure,
concentration information sur les premiers facteurs
Répartition homogène "brui t" et information

7/22/2019 chimiomtrie2
http://slidepdf.com/reader/full/chimiomtrie2 11/18
11
seuls utilisés. Or le bruit reste également réparti pour tous les facteurs: l'information "abandonnée" n'est donc
pratiquement que du bruit, et on a donc globalement une réduction de celui-ci.
On élimine ainsi les inconvénients et on ajoute les avantages des 2 précédentes méthodes:
-ILS: Le nombre de longueurs d'onde utilisées n'est limité que par la puissance de calcul. Il n'y a plus de
problèmes de colinéarité puisqu'on ne prend que des combinaisons d’absorbances orthogonales entre elles. Le nombre
de facteurs doit toutefois être inférieur ou égal au nombre d'échantillons.
-MLR : Il n'y a pas de problème d'impuretés puisque le prédicteur des concentrations est déterminé par ILS à
partir des scores, donc de façon indépendante pour chaque composé.
Dans les 2 cas, on réduit le risque d'"overfitting" (modélisation du bruit) en réduisant le nombre de variables.
Il existe principalement 2 méthodes d'analyse des facteurs:
2.3.1. PCR (Principal Component Régression)
Cette méthode utilise la matrice des covariances des absorbances centrées, A'A. Elle part du principe que pour
choisir une nouvelle base de variables indépendantes, il suffit, par définition, que les covariances de ces variables entre
elles soient nulles. La matrice de changement de base est donc la matrice qui va rendre la matrice de covariance
diagonale. Elle est donc construite à partir des vecteurs propres de A'A. Les valeurs propres représentent alors la
variance des scores, et donc leur contribution à la modélisation: Pour effectuer la réduction de données, il suffit alors de
ne conserver que les h scores correspondant aux plus fortes valeurs propres.
σ
σ
σ
σ
σ
σ
2
1 1
1
2
2
1
2
2
1
2
0
0
0
0
( ) ( , )
( , ) ( )
( )
( )
( )
( )
a Cov a a
Cov a a a
t
t
t
t
n
n n n h
L
M O M
L
L
M O M
L
L
M O M
L
→
→
h ≤≤ n
Cette méthode est très efficace, mais elle ne tient pas compte dans la première phase d’étalonnage, (choix de la
nouvelle base) des informations concentration, qui ne sont utilisées que dans la phase de régression des concentrations
sur les scores.
Il peut pourtant arriver que des variations importantes des absorbances ne soient absolument pas corrélés avec les
concentrations.
On lui préfère donc souvent une méthode plus récente et plus complexe:
2.3.2. PLS (Partial Least Squares): Moindres carrés partiels
Cette méthode, plus récente (1980), consiste à construire ensembles les matrices de changement de base W
("Loadings Vectors") et de prédiction V en utilisant conjointement les absorbances et les concentrations.
Nous n'expliquerons que très schématiquement l'algorithme le plus simple (dit « non orthogonal ») pour 1 seul
composant (PLS1). Pour une explication plus complète ou pour l'algorithme avec plusieurs composants (PLS2), on se
reportera à la littérature (1).
Etalonnage PLS:
A0 et C0 sont les valeurs initiales d'absorbance et de concentration normées et centrées.
A : matrice m lignes, k colonnes
C : vecteur (1 seul composé) m valeurs
(1) : Rechercher, en régressant A0 sur C0, la composante de A la plus corrélée avec les variations de concentration:
A0 = C0W1 + E
m : nombre d’échantillons d’étalonnage
k : nombre d’Xvariables (ou de longueurs d’ondes)

7/22/2019 chimiomtrie2
http://slidepdf.com/reader/full/chimiomtrie2 12/18
12
W1 sera alors la première composante de la matrice de changement de base W. C'est l'opérateur "projecteur
moyen" de A0 sur C0 (et qui ne projettera donc probablement pas parfaitement chaque composante de A0 sur C0)
W1 = (C'0C0)-1C'0A0
W1C0 est donc la projection de A0 sur C0 dans l'espace des échantillons.
W1 est alors normalisée: On a alors W 1W'1 = Id
(2) : Rechercher le "score" (projeté) correspondant à ce "projecteur moyen" en régressant A 0 sur W1:
A0 = T1W1 +E
donc T1 = A0W'1(W1W'1)-1 = A0W1
(3): Rechercher en régressant C0 sur T1 la composante du prédicteur V1 correspondant à ce facteur:
(On fait alors une ILS).
C = T1v1 + E (1 compo --> v1 scalaire)
Donc v1 = (T'1T1)-1T'1C
(4) : Calculer les résidus d'absorbance et de concentration (information non utilisée, orthogonale à la précédente) en
retranchant l'information déjà modélisée:
A1 = A0 - T1W1
C1 = C0 - T1v1
(6) : Reprendre en (1) avec ces nouvelles valeurs et continuer à construire W et V jusqu'à atteindre le nombre de
facteurs souhaité.
Il s'agit donc d'une méthode itérative (1 itération par facteur), pour chaque itération, l'information utilisée pour
construire le modèle (info A ou C) est retranchée pour l'itération suivante.
Apres les n itérations, l'information (A ou C) restante est appelée résidu (d'absorbance ou de concentration).
Prédiction PLS: Il s'agit pratiquement de l'opération inverse: On extrait de A successivement les informations
correspondant à chaque facteur avec lesquelles on construit la concentration C (initialement nulle), soit, pour chaque
facteur d'indice a:
(1) : Initialisation : A0 normée centrée , C0=0
(2) : Calcul de la contribution de ce score à la concentration:
ca = ca-1 + Tava
(3) : Calcul du nouveau résidu d'absorbance:
Aa+1 = Aa- TaWa
(4) : Continuer en (2) avec le nouveau résidu d'absorbance jusqu'à atteindre le nombre de facteurs souhaités.
La concentration finale, après décentrage et dénormage, sera donc la somme des contributions des différents
facteurs.
7/22/2019 chimiomtrie2
http://slidepdf.com/reader/full/chimiomtrie2 13/18
13
1
2
3
4
5
1
2
3
4
5
Absorbances Concentrations
Modèle
Résidus
1
2
3
4
5
Absorbances
Modèle Concentrations
Résidus
1
2
3
4
5
Absorbances
Modèle Concentrations
Résidus
Même ordr ede grandeur? Même ordr ede grandeur?
M O D E L I S A T I O N
P R E D I C T I O N
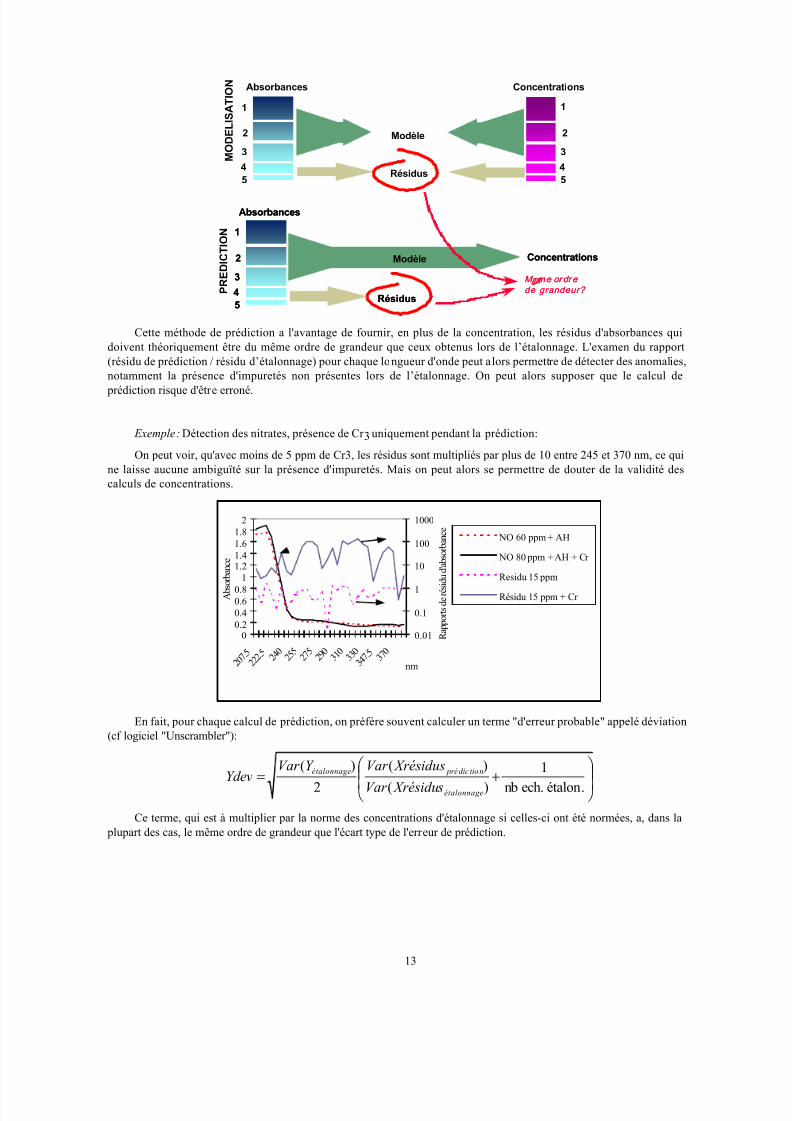
Cette méthode de prédiction a l'avantage de fournir, en plus de la concentration, les résidus d'absorbances qui
doivent théoriquement être du même ordre de grandeur que ceux obtenus lors de l’étalonnage. L'examen du rapport
(résidu de prédiction / résidu d’étalonnage) pour chaque longueur d'onde peut alors permettre de détecter des anomalies,
notamment la présence d'impuretés non présentes lors de l’étalonnage. On peut alors supposer que le calcul de
prédiction risque d'être erroné.
Exemple: Détection des nitrates, présence de Cr 3 uniquement pendant la prédiction:
On peut voir, qu'avec moins de 5 ppm de Cr3, les résidus sont multipliés par plus de 10 entre 245 et 370 nm, ce qui
ne laisse aucune ambiguïté sur la présence d'impuretés. Mais on peut alors se permettre de douter de la validité des
calculs de concentrations.
00.2
0.4
0.6
0.8
1
1.21.4
1.61.8
2
2 0 7. 5
2 2 2. 5
2 4 0
2 5 5
2 7 5
2 9 0
3 1 0
3 3 0
3 4 7. 5
3 7 0
nm
A b s o r b a n c e
0.01
0.1
1
10
100
1000
R a p p o r t s d e r é s i d u d ' a b s o r b a n c
e
NO 60 ppm + AH
NO 80 ppm + AH + Cr
Residu 15 ppm
Résidu 15 ppm + Cr
En fait, pour chaque calcul de prédiction, on préfère souvent calculer un terme "d'erreur probable" appelé déviation
(cf logiciel "Unscrambler"):
+=
.étalonech.nb
1
)(
)(
2
)(
étalonnage
prédic tionétalonnage
XrésidusVar
XrésidusVar Y Var Ydev
Ce terme, qui est à multiplier par la norme des concentrations d'étalonnage si celles-ci ont été normées, a, dans la
plupart des cas, le même ordre de grandeur que l'écart type de l'erreur de prédiction.
7/22/2019 chimiomtrie2
http://slidepdf.com/reader/full/chimiomtrie2 14/18
14
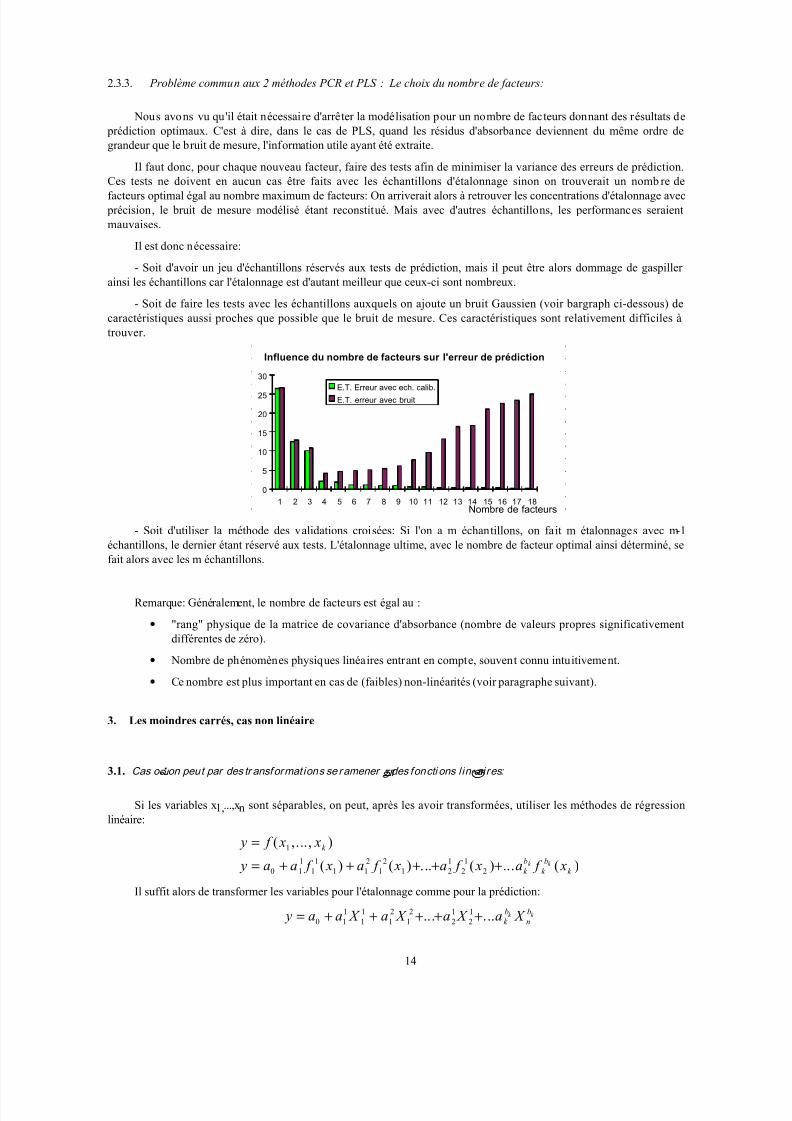
2.3.3. Problème commun aux 2 méthodes PCR et PLS : Le choix du nombre de facteurs:
Nous avons vu qu'il était nécessaire d'arrêter la modélisation pour un nombre de facteurs donnant des résultats de
prédiction optimaux. C'est à dire, dans le cas de PLS, quand les résidus d'absorbance deviennent du même ordre de
grandeur que le bruit de mesure, l'information utile ayant été extraite.
Il faut donc, pour chaque nouveau facteur, faire des tests afin de minimiser la variance des erreurs de prédiction.
Ces tests ne doivent en aucun cas être faits avec les échantillons d'étalonnage sinon on trouverait un nomb re defacteurs optimal égal au nombre maximum de facteurs: On arriverait alors à retrouver les concentrations d'étalonnage avec
précision, le bruit de mesure modélisé étant reconstitué. Mais avec d'autres échantillons, les performances seraient
mauvaises.
Il est donc nécessaire:
- Soit d'avoir un jeu d'échantillons réservés aux tests de prédiction, mais il peut être alors dommage de gaspiller
ainsi les échantillons car l'étalonnage est d'autant meilleur que ceux-ci sont nombreux.
- Soit de faire les tests avec les échantillons auxquels on ajoute un bruit Gaussien (voir bargraph ci-dessous) de
caractéristiques aussi proches que possible que le bruit de mesure. Ces caractéristiques sont relativement difficiles à
trouver.
10.744.2
4.55
4.85
5.16
5.48
6
7.7
9.6
13.3
16.4
16.8
21
Influence du nombre de facteurs sur l'erreur de prédiction
0
5
10
15
20
25
30
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
E.T. Erreur avec ech. calib.
E.T. erreur avec bruit
Nombre de facteurs
- Soit d'utiliser la méthode des validations croisées: Si l'on a m échantillons, on fait m étalonnages avec m-1échantillons, le dernier étant réservé aux tests. L'étalonnage ultime, avec le nombre de facteur optimal ainsi déterminé, se
fait alors avec les m échantillons.
Remarque: Généralement, le nombre de facteurs est égal au :
• "rang" physique de la matrice de covariance d'absorbance (nombre de valeurs propres significativement
différentes de zéro).
• Nombre de phénomènes physiques linéaires entrant en compte, souvent connu intuitivement.
• Ce nombre est plus important en cas de (faibles) non-linéarités (voir paragraphe suivant).
3. Les moindres carrés, cas non linéaire
3.1. Cas où on peut par des tr ansformat ions se ramener àdes foncti ons l inéaires:
Si les variables x1,...,xn sont séparables, on peut, après les avoir transformées, utiliser les méthodes de régression
linéaire:
y f x x
y a a f x a f x a f x a f x
k
k
b
k
b
k k k
=
= + + + + +
( ,..., )
( ) ( ) ... ( ) ... ( )
1
0 1
1
1
1
1 1
2
1
2
1 2
1
2
1
2
Il suffit alors de transformer les variables pour l'étalonnage comme pour la prédiction:
y a a X a X a X a X k
b
n
bk k = + + + + +0 1
1
1
1
1
2
1
2
2
1
2
1... ...

7/22/2019 chimiomtrie2
http://slidepdf.com/reader/full/chimiomtrie2 15/18
15
Cas particulier classique: la régression polynomiale: une seule variable x remplacée par n variables x1...xn:
y a a x a x a x
y a a X a X a X
n
n
n n
= + + + += + + + +
0 1 2
2
0 1 2 2
...
...
Cette méthode peut s’appliquer aussi aux Yvariables.
3.2. PL S peu t modéliser des phénomènes non l inéai res :
Bien que cela puisse paraître paradoxal pour une méthode basée sur l’algèbre linéaire, PLS (tout comme PCR) peut
modéliser des phénomènes non linéaires.
En pratique, si différentes Xvariables ont des non-linéarités « différentes » dans leur relation avec les Yvariables,
alors la modélisation PLS peut implicitement combiner ces différentes non-linéarités afin d’exprimer une relation linéaire
entre X et Y, et cela sans terme supplémentaire « non linéaire ».
Mais si toutes les relations entre X et Yvariables ont le même type de non-linéarité alors la modélisation rigoureuse
est impossible. Il est souvent possible de s’en rapprocher en ajoutant des termes fonctions non linéaires des Xvariables
comme Xvariables supplémentaires, par conséquent linéairement indépendantes des anciennes (c’est ce qui est fait en
régression polynomiale).
Toutefois, cela se fait toujours au prix d’une complexification du modèle (augmentation du nombre de facteurs), et il
est toujours préférable, lorsque cela est possible, de faire un traitement de linéarisation des X et/ou des Yvariables (c’est
ce que l’on fait implicitement en spectrophotométrie en transformant les intensités lumineuses en absorbances).
3.3. Méthodes itérat ives, ou méthodes basées sur des opt imi sations :
Ces méthodes, peu utilisées en chimiométrie sortent du cadre de cet ouvrage, à une exception notable, les réseaux
neuronaux, traités dans le paragraphe suivant.
4. Réseaux neuronaux
Les réseaux neuronaux peuvent être considérés comme un algorithme d'analyse multivariable. C’est en fait un
modèle de comportement universel dans la mesure où il n’est plus nécessaire de faire d’hypothèse mathématique
initiale. Seule la structure du réseau peut changer (nombre et taille des couches intermédiaires, fonctions de transfert).
Variables
d'entréeRESEAU
Variables
de sortie
(= cibles)

7/22/2019 chimiomtrie2
http://slidepdf.com/reader/full/chimiomtrie2 16/18
16
4.1. Structure général e du réseau:
Entrées Sorties Cibles
couche
intermédiaire
W2W1
B1
B 2Offsets:
Coef.
s na ti ues
Fonction de
Transfert non linéaire
1 1, ,*
k k
i
ii B W E + ∑
k
Le réseau de neurones est composé d’une « couche d’entrée » correspondant à l’Xvariable à transformer, d’une couche
de sortie fournissant l’Yvariable modélisée et éventuellement d’une ou plusieurs couches intermédiaires, de tailles
choisies par le modélisateur. Les couches de sortie et intermédiaires sont des combinaisons linéaires de la couche qui les
précède, les coefficients W i,j étant appelés « coefficients synaptiques ». On ajoute parfois un terme constant Bi appelé
« offset ». Ce qui fait la richesse des réseau de neurones c’est la transformation de cette combinaison linéaire par une
fonction de transfert généralement non linéaire (souvent une fonction sigmoïde, ou bien linéaire tronquée).
On peut remarquer que si la fonction de transfert est linéaire, alors le réseau est équivalent aux méthodes linéaires
décrites précédemment, les couches intermédiaires correspondant aux facteurs. Seul le mode de calcul des coefficients,
itératif et appelé apprentissage ici, est différent.
4.2. Appr enti ssage ( = étal onnage):
On utilise, comme pour toute analyse multivariable, 2 jeux de variables:
- Variables d'entrée (Xvariables)
- Variables cibles (Yvariables).
Le but est alors de trouver, par approximations successives les coefficients synaptiques (W) et les offsets (B) telsque les cibles et les sort ies soient les plus proches possibles au sens des moindres carrés : On va donc encore optimiser
la somme des carrés des erreurs (écart cible – sortie)
C'est un problème d'optimisation qui se résout par itérations. Si les fonctions de transfert sont linéaires, alors le
problème peut se ramener à un problème d'analyse multivariable classique (simple ou à analyse de facteurs si il y a une
couche de neurones intermédiaires).
Sinon, le procédé d'optimisation le plus utilisé est la rétro-propagation:
Il consiste à propager les erreurs (différences entre la sortie et la cible correspondante) de la sortie vers l'entrée en
modifiant « légèrement » au passage les coefficients W et B dans le sens d’une diminution de l’erreur. La méthode est
appliquée avec tout le jeu d'échantillons un grand nombre de fois jusqu'à obtention du degré de convergence
"souhaité" .
Il n’est pas nécessaire de faire converger le réseau outre mesure: On risque en effet alors de faire de la sur-
modélisation qui, comme cela a été expliqué dans le paragraphe traitant du linéaire, peut être néfaste pour la
prédiction.

7/22/2019 chimiomtrie2
http://slidepdf.com/reader/full/chimiomtrie2 17/18
17
Il est donc conseillé de tester le réseau, en cours de convergence, avec un jeu d’échantillons non utilisés pour
l’apprentissage.
4.3. Prédiction:
On peut alors "exciter » le réseau (W,B) avec de nouvelles Xvariables d'entrée afin de trouver les Yvariables de
sortie correspondantes (à priori inconnues).
Utilisations:
Essentiellement reconnaissance de formes (au sens large):
* spectres, courbes (--> chimiométrie), systèmes multicapteurs.
* images, caractères
* voix, sons...
mais aussi:
* approximation de fonctions
* systèmes experts, intelligence artificielle
5. Conclusion
La chimiométrie, ou analyse multivariable, permet de construire un modèle de comportement de phénomènes
physico-chimiques afin d’exploiter ces phénomènes en instrumentation. Sa principale difficulté, une fois les méthodes
comprises, consiste à adapter la complexité du modèle aux qualités métrologiques des mesures, et donc de trouver un
compromis entre précision et fiabilité de ce modèle.
D’une façon générale, alors que les méthodes d’analyse de facteurs semblent plus performantes pour modéliser les
phénomènes linéaires ou peu non linéaires, les réseaux neuronaux tirent leur épingle du jeu pour les phénomènes
fortement non linéaires qu’ils sont capables de modéliser de manière moins complexe.
6. Annexe: Rappels d'algèbre linéaire
* On appelle matrice A un ensemble de m*n scalaires disposés en m colonnes de n nombres
( ) ( ) A
a a
a a
a B
b b
b b
b
n
m
n
m
k
i
l
n
l
m
j
k =
= =
=1
1 1
1
1
1 1
1
L
M O M
L
L
M O M
L
( )α β α β A A a a
A B a b
k
i
k
i
k
i
j
k
i
+ = +
=
∑
' '
*
Transposée: ( ) ( ) A a A ak
i
i
k = → ='
Soit à résoudre A = C*X, où X est l'inconnue:
Si C est une matrice carrée, (nombre de lignes = nombre de colonnes), on appelle matrice inverse de C la matrice C-1
telle que:
C C C C Id * *− −
= = =
1 1
1 0 0
0 00 0 1
O

7/22/2019 chimiomtrie2
http://slidepdf.com/reader/full/chimiomtrie2 18/18
18
C-1 n'existe que si les colonnes de C sont linéairement indépendantes.
alors X = C-1*A
Si C est non carrée (n lignes, m colonnes), on peut écrire:
C'*A = C'*C*X
C'C est alors carrée (n * n) et peut-être inversible si ses colonnes sont linéairement indépendantes ce qui impose
que nRRm. (Si 2 colonnes de C sont colinéaires alors C’C ne sera aussi pas inversible). On peut alors écrire:
X = (C'*C)-1*C'*A, X est appelé pseudo-inverse.
On appelle vecteur une matrice à une seule colonne.
On appelle vecteur propre d'une matrice C carrée (n * n) un vecteur V tel que:
C * Va = r a*Va
Les différentes valeurs scalaires r 1...r n sont appelés valeurs propres de C. Elles sont uniques alors que le vecteur
propre est défini à une constante près. Une condition nécessaire et suffisante pour que C soit inversible ( = non
singulière) est que ses valeurs propres soient toutes différentes de 0.On appelle base C1 un ensemble de n1 vecteurs linéairement indépendants pouvant servir de repère dans un espace
de dimension n1. Lorsqu' on change de base C1 à C2 (n2<=n1) , le passage des anciennes coordonnées X1 aux nouvelles
coordonnées X2 se fait à l'aide d'une matrice de changement de base M qui est la matrice de projection de X1 sur X2:
X2 = M*X1
7. Bibliographie
1: "Multivariate Calibration" Harald Martens, Tormod Naes ed: John Wiley & sons Chichester
2: " Practical Guide to Chemometrics" Stephen John Haswell ed: Marcel Dekker, Inc New York
3: "Multivariate Statistitcal Methods" A. Primer, Bryan F.J. Manly ed: Chapman & Hall London
4: Techniques de l'ingénieur: optimisation
5: Logiciel MATLAB (distribué par Scientific Software 92 Sèvres) et modules:
- Chemometrics
- Optimization
- Neural network
6: Logiciel Unscrambler (Camo, Lolav Tryggvasons gt.24, N-7011 Trondheim - Norvège)
7 :« La régression PLS, théorie et pratique » M. Tennenhaus ed : Technip, Paris
Sites Internet:
Galactic algorithmes : http://www.galactic.com/Algorithms/default.asp
Chemometrics World - John Wiley & Sons, Ltd.: http://www.wiley.co.uk/wileychi/chemometrics/
Chemometrics (Martin Huehne) : http://tmec.nectec.or.th/thfi/chemom.htm
Homepage of chemometrics info on multivariate calibration data analysis ) :
http://www.acc.umu.se/~tnkjtg/chemometrics/index.html
Chemometrics Online (modélisation en ligne avec vos données, inscription gratuite) :http://chemometrics.odu.edu


















