
Chapitre 2 : Matériaux de l’étude et principaux...
30
Chapitre 2 : Matériaux de l’étude et principaux moyens de caractérisation mis en œuvre
Transcript of Chapitre 2 : Matériaux de l’étude et principaux...

Chapitre 2 : Matériaux de l’étude et principaux moyens de caractérisation mis en œuvre

Chapitre 2 : Matériaux de l’étude et principaux moyens de caractérisation mis en œuvre
70
Sommaire
I. INTRODUCTION __________________________________________________________ 71
II. PRÉSENTATION DU SYSTÈME ROULEMENT À BILLES ______________________ 71
III. CHOIX DES MATÉRIAUX DE L’ÉTUDE______________________________________ 73
III.1. Présentation des matériaux ________________________________________________ 73 III.1.1. Effet des charges (à matrice constante) ______________________________________________ 73
III.1.2. Effet de la matrice (à charges constantes) ____________________________________________ 74
III.1.3. Effet du taux de réticulation _______________________________________________________ 74
III.2. Données préliminaires sur les matériaux _____________________________________ 75
III.3. Méthodes de préparation des matériaux _____________________________________ 78
IV. DIFFÉRENTES TECHNIQUES EXPÉRIMENTALES UTILISÉES ________________ 79 IV.1. Caractérisations tribologiques _____________________________________________ 79
IV.1.1. Bancs d’essai SNR ______________________________________________________________ 79
IV.1.2. Tribomètre bille/plan ____________________________________________________________ 80
IV.2. Caractérisations mécaniques_______________________________________________ 82 IV.2.1. Spectrométrie mécanique _________________________________________________________ 82
IV.2.2. Essais grandes déformations – Vidéotraction__________________________________________ 86
IV.2.3. La Nanoindentation _____________________________________________________________ 87
IV.3. Caractérisations physico-chimiques _________________________________________ 93 IV.3.1. Analyse ThermoGravimétrique (ATG) ______________________________________________ 93
IV.3.2. Analyse Thermique Différentielle (ATD) ____________________________________________ 93
IV.3.3. Mesure des énergies de surface – Méthode de la goutte posée ____________________________ 93
IV.4. Caractérisations microstructurales _________________________________________ 96 IV.4.1. La Microscopie_________________________________________________________________ 96
IV.4.2. ESCA (Electron Spectroscopy for Chemical Analysis) __________________________________ 96
RÉFÉRENCES BIBLIOGRAPHIQUES ____________________________________________ 98

Chapitre 2 : Matériaux de l’étude et principaux moyens de caractérisation mis en œuvre
71
I. Introduction
Comme on a pu le constater dans la première partie de l’étude bibliographique, les
élastomères chargés utilisés dans l’industrie ont des compositions chimiques très complexes.
Leurs propriétés mécaniques ainsi que leur comportement tribologique sont directement reliées
aux différents composants utilisés dans la préparation des matériaux. Ce chapitre propose donc,
dans un premier temps, une présentation rapide des matériaux de l’étude, ainsi qu’une analyse des
différentes données qui nous ont été fournies par la société Paulstra (élaborateur).
II. Présentation du système roulement à billes
Un roulement à billes est un mécanisme destiné à diminuer les frottements entre les pièces
roulant l’une sur l’autre, et qui est constitué de billes insérées entres deux organes flottants. La
fonction première de ce système mécanique est de faciliter la rotation et d’abaisser le coefficient
de frottement entre deux organes. Une deuxième fonction est d’augmenter la tenue à l’usure du
système puisque que le roulement permet de diminuer les points de contacts entre deux éléments
mécaniques, type moyeu-roue.
Un roulement est constitué de billes disposées dans des cages et roulant entre la bague intérieure
et la bague extérieure en présence de graisse. Pour protéger ces corps roulant de l’intrusion de
particules extérieures, des joints d’étanchéité sont ajoutés de chaque côté du roulement, entre les
bagues intérieure et extérieure (cf. Figure 1).
Joint d ’étanchéité
Bague intérieure
Billes: corps roulant
Bague extérieure
Cage
Figure 1 : Différents composants d’un roulement à billes

Chapitre 2 : Matériaux de l’étude et principaux moyens de caractérisation mis en œuvre
72
Le système joint d’étanchéité est la partie du roulement à billes qui nous intéresse dans
cette étude (cf. Figure 2). Il est constitué d’une armature en métal, recouverte d’élastomère. Les
parties critiques d’un joint sont les lèvres en élastomère qui viennent s’appuyer sur un déflecteur
en acier inoxydable, assurant ainsi la fonction d’étanchéité (cf. Figure 3. Au cours de l’utilisation,
déflecteur et lèvres sont en rotation l’un par rapport à l’autre, avec de la graisse à l’interface
(présente initialement dans l’espace entre les deux lèvres). De plus, des particules abrasives
extérieures (projections provenant de la route par exemple), nommées ‘pollution’, sont
susceptibles de pénétrer dans le joint, et ainsi dégrader le système. Lorsque cette polllution
pénètre dans les corps roulant, le roulement devient inutilisable, car les billes sont endommagées
et le système global pert de son efficacité.
Figure 2 : Roulement à billes ; zoom sur un joint d’étanchéité
Déflecteurmétallique
Lèvres en élastomère
Armaturemétallique
Entrée de pollution
Graisse
BEBI Figure 3 : Schéma d’un joint d’étanchéité (BI : bague intérieure ; BE :bague extérieure)
Chapitre 2 : Matériaux de l’étude et principaux moyens de caractérisation mis en œuvre
73
III. Choix des matériaux de l’étude
Plusieurs séries de matériaux nous ont été fournies par la société Paulstra, chacune d’entre
elles répondant à la variation d’un paramètre nous semblant intéressant pour la suite de l’étude.
Ainsi, nous avons pu étudier l’effet des charges, l’effet de la composition de la matrice et l’effet
du taux de réticulation. Cependant, pour des raisons de confidentialité, il ne nous est pas possible
de fournir plus de détails quant à la composition précise de ces matériaux ; si ce n’est qu’il s’agit,
dans tous les cas, d’une matrice Acrylonitrile-Butadiène (NBR), renforcée par des charges et dont
la composition comporte également de nombreux composants, tels que ceux cités dans le
Chapitre 1.
De tous ces lots de matériaux, on peut sortir un dénominateur commun qui sert de base : le
matériau A_240. C’est le matériau qui se rapproche le plus, en terme de composition globale, des
matériaux standards utilisés dans la fabrication des joints.
III.1. Présentation des matériaux
III.1.1 Effet des charges (à matrice constante)
On a pu voir dans l’étude bibliographique que les charges jouaient un grand rôle tant du
point de vue propriétés mécaniques que du comportement tribologique. Il nous a semblé donc
pertinent de baser une partie de cette étude sur des matériaux présentant des variations en terme
de charges. Un tableau de la composition des différents matériaux en terme de charges est
représenté ci-dessous.
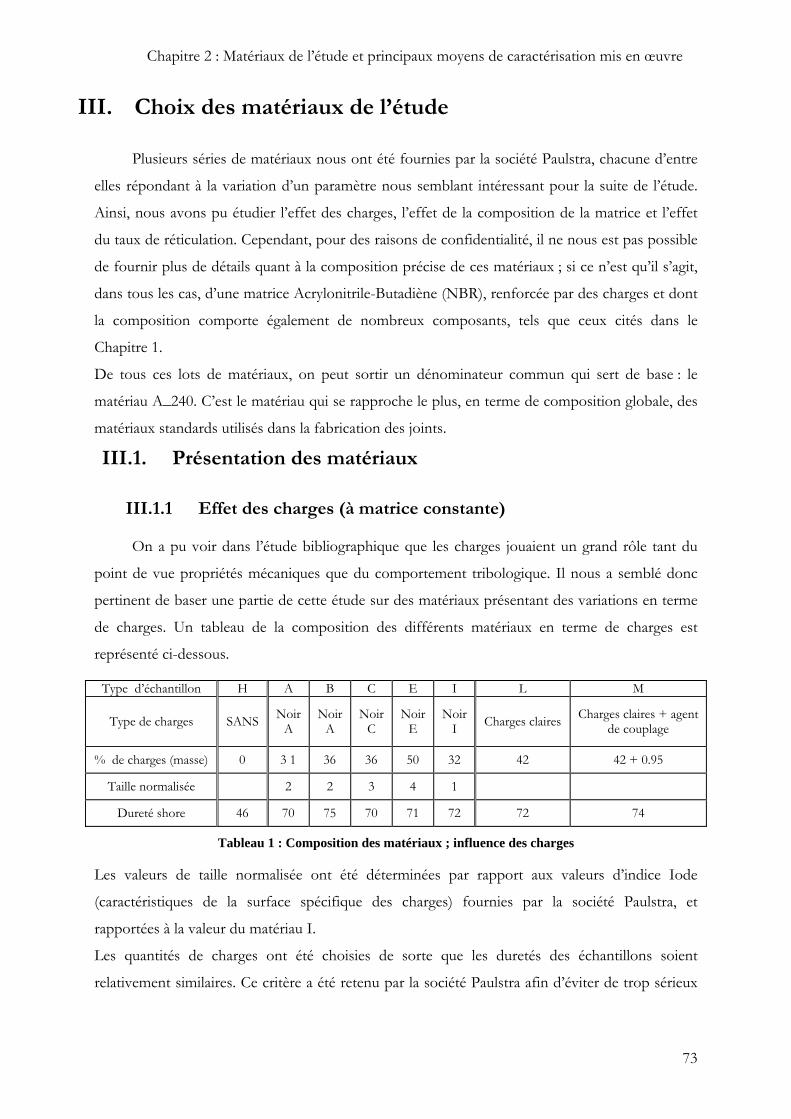
Type d’échantillon H A B C E I L M
Type de charges SANS Noir A
Noir A
Noir C
Noir E
Noir I Charges claires Charges claires + agent
de couplage
% de charges (masse) 0 3 1 36 36 50 32 42 42 + 0.95
Taille normalisée 2 2 3 4 1
Dureté shore 46 70 75 70 71 72 72 74
Tableau 1 : Composition des matériaux ; influence des charges
Les valeurs de taille normalisée ont été déterminées par rapport aux valeurs d’indice Iode
(caractéristiques de la surface spécifique des charges) fournies par la société Paulstra, et
rapportées à la valeur du matériau I.
Les quantités de charges ont été choisies de sorte que les duretés des échantillons soient
relativement similaires. Ce critère a été retenu par la société Paulstra afin d’éviter de trop sérieux

Chapitre 2 : Matériaux de l’étude et principaux moyens de caractérisation mis en œuvre
74
problèmes de mise en œuvre ; mais aussi et surtout, pour avoir des différences de propriétés ne
compromettant pas la réalisations des essais tribologiques.
Comme on peut le voir dans le Tableau 1, ces matériaux ont été retenus pour identifier le rôle des
charges à plusieurs niveaux :
∑ Rôle de la taille des charges : comparaison des échantillons B/C et aussi A/I.
∑ Rôle du taux de charges : comparaison entre A et B, et aussi entre A-I et E.
∑ Nature des charges : comparaison A-E-I et L., et L/M pour l’agent de couplage.
III.1.2 Effet de la matrice (à charges constantes)
Dans le but d’étudier l’influence des propriétés dissipatives sur l’usure, un nouveau
matériau, toujours basé sur le A_240, a été utilisé pour fabriquer des joints et des plaques de
matériaux.
Ce matériau, nommé 0_240, présente une composition de matrice différente par rapport à notre
base ; c’est-à-dire que le taux d’acrylonitrile (ACN) a été augmenté, afin d’élever la température de
transition vitreuse Tg (∆Tg≈15°C). Concernant les autres paramètres caractéristiques de nos
élastomères chargés, ils sont restés identiques à la base : même composition (antioxydants…),
avec surtout même charges (nature, taille et taux), et ils se situent tous les deux à leur optimum de
vulcanisation.
Tv (185°C) Tg (°C) %ACN*A_240 240s -28O_240 240s -13
∆%=10
Tableau 2 : Caractéristiques des matériaux A_240 et O_240
* La relation entre la différence de Tg et la différence de taux d’ACN vérifie la loi de Gordon-Taylor évoquée dans la partie bibliographique.
III.1.3 Effet du taux de réticulation
Une autre possibilité est d’analyser les différences de comportement tribologique pour des
matériaux présentant des réticulations plus ou moins complètes. En effet, on a vu dans la partie
bibliographie, que les élastomères pouvaient évoluer sous l’effet d’une contrainte, d’une
température élevée ou d’une combinaison des deux, et présenter une post-réticulation.
Pour ce faire, en se basant sur le matériau A_240, un jeu de matériaux présentant des temps de
vulcanisation (Tv) plus faibles, et donc des réticulations incomplètes, a été préparé. Nos
matériaux A_120 et A_150 sont présentés dans le tableau suivant.

Chapitre 2 : Matériaux de l’étude et principaux moyens de caractérisation mis en œuvre
75
Tv (à 185°C) gonflement Tg
A_240 240 s 1,82 -28°C
A_150 150 s 1,85 -34°C
A_120 120 s 1,89 -33°C Tableau 3 : Caractéristiques des matériaux avec taux de réticulation différents (A_120, A_150, A_240)
La valeur du gonflement donnée dans le tableau ci-dessus est une indication du taux de
réticulation de chaque matériau ; plus le gonflement est important, moins la réticulation est
importante. Dans notre cas, les différences sont faibles, et donc les matériaux sont assez proches.
III.2. Données préliminaires sur les matériaux
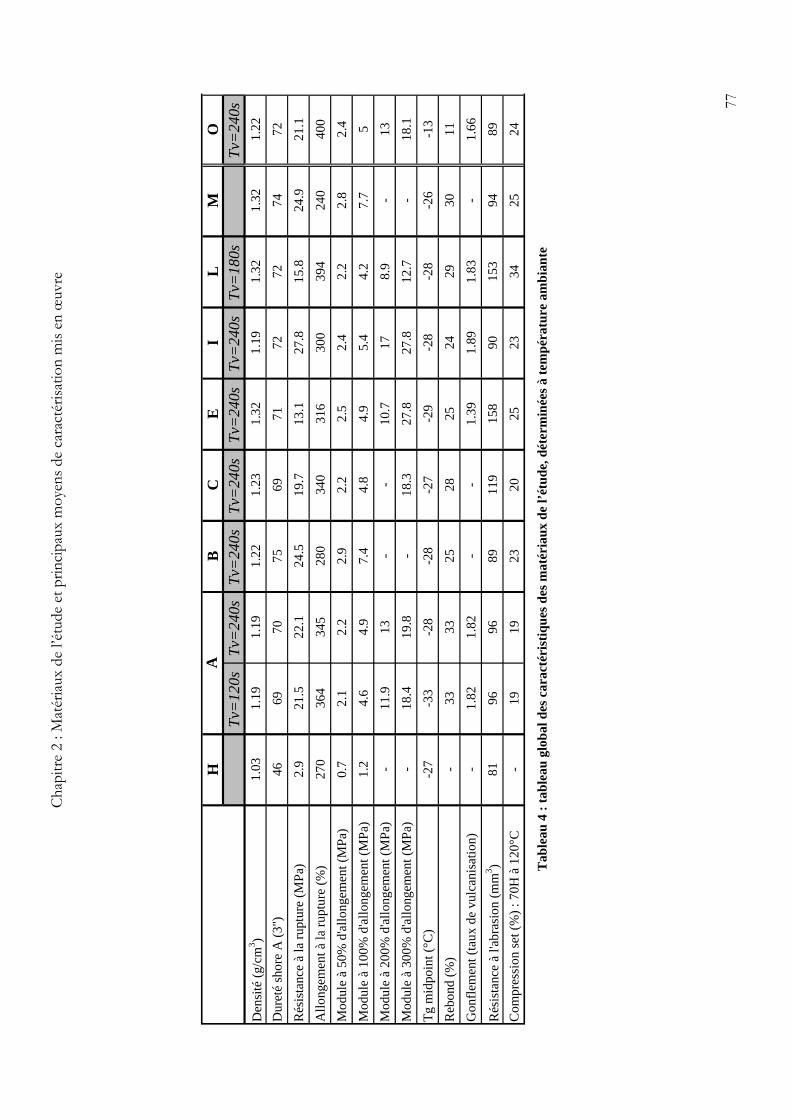
Les principales données fournies par la société Paulstra concernant les matériaux sont
regroupées dans un tableau global donné ci-dessous (cf. Tableau 4).
Des tests de traction simple (à une vitesse de sollicitation donnée et à température
ambiante) ont permis de déterminer les modules sécants à différents allongements ainsi que les
propriétés à la rupture des matériaux étudiés.
L’évolution des valeurs de module sécant avec l’allongement est représentée sur la Figure 4
pour les 3 matériaux A (A_120, A_150 et A_240) et le matériau O_240. Ainsi, on ne peut
remarquer que peu d’écart en terme de module (≈5%) entre les matériaux A, et cet écart
augmente légérement en absolu avec l’augmentation d’allongement. Aux grandes déformations,
on retrouve tout de même bien un classement en accord avec le gonflement (le A_240, étant le
plus réticulé, possède un module légèrement supérieur). Au niveau de la comparaison entre le
A_240 et le O_240, une fois encore les écarts sont faibles, sauf aux grandes déformations où
l’ordre s’inverse, probablement du fait d’un consolidation moins importante pour le O_240.
0
4
8
12
16
20
0 50 100 150 200 250 300 350% allongement
mod
ule
séca
nt (M
Pa)
A_120A_150A_240O_240
Figure 4 : évolution du module sécant en fonction de l’allongement pour les matériaux A et O
Chapitre 2 : Matériaux de l’étude et principaux moyens de caractérisation mis en œuvre
76
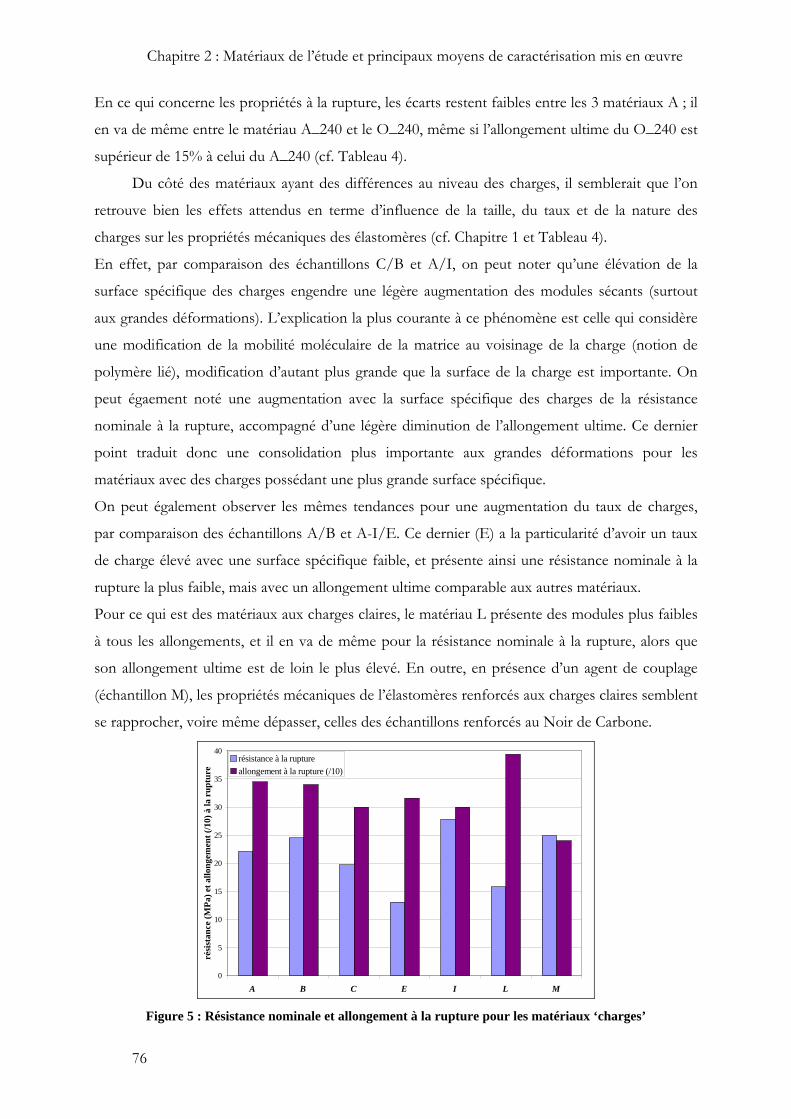
En ce qui concerne les propriétés à la rupture, les écarts restent faibles entre les 3 matériaux A ; il
en va de même entre le matériau A_240 et le O_240, même si l’allongement ultime du O_240 est
supérieur de 15% à celui du A_240 (cf. Tableau 4).
Du côté des matériaux ayant des différences au niveau des charges, il semblerait que l’on
retrouve bien les effets attendus en terme d’influence de la taille, du taux et de la nature des
charges sur les propriétés mécaniques des élastomères (cf. Chapitre 1 et Tableau 4).
En effet, par comparaison des échantillons C/B et A/I, on peut noter qu’une élévation de la
surface spécifique des charges engendre une légère augmentation des modules sécants (surtout
aux grandes déformations). L’explication la plus courante à ce phénomène est celle qui considère
une modification de la mobilité moléculaire de la matrice au voisinage de la charge (notion de
polymère lié), modification d’autant plus grande que la surface de la charge est importante. On
peut égaement noté une augmentation avec la surface spécifique des charges de la résistance
nominale à la rupture, accompagné d’une légère diminution de l’allongement ultime. Ce dernier
point traduit donc une consolidation plus importante aux grandes déformations pour les
matériaux avec des charges possédant une plus grande surface spécifique.
On peut également observer les mêmes tendances pour une augmentation du taux de charges,
par comparaison des échantillons A/B et A-I/E. Ce dernier (E) a la particularité d’avoir un taux
de charge élevé avec une surface spécifique faible, et présente ainsi une résistance nominale à la
rupture la plus faible, mais avec un allongement ultime comparable aux autres matériaux.
Pour ce qui est des matériaux aux charges claires, le matériau L présente des modules plus faibles
à tous les allongements, et il en va de même pour la résistance nominale à la rupture, alors que
son allongement ultime est de loin le plus élevé. En outre, en présence d’un agent de couplage
(échantillon M), les propriétés mécaniques de l’élastomères renforcés aux charges claires semblent
se rapprocher, voire même dépasser, celles des échantillons renforcés au Noir de Carbone.
0
5
10
15
20
25
30
35
40
A B C E I L M
rési
stan
ce (M
Pa) e
t allo
ngem
ent (
/10)
à la
rup
ture
résistance à la ruptureallongement à la rupture (/10)
Figure 5 : Résistance nominale et allongement à la rupture pour les matériaux ‘charges’
Chap
itre
2 : M
atér
iaux
de l’
étud
e et
prin
cipau
x m
oyen
s de
cara
ctér
isatio
n m
is en
œuv
re
77
HB
CE
IL
MO
Tv=
120s
Tv=
240s
Tv=
240s
Tv=
240s
Tv=
240s
Tv=
240s
Tv=
180s
Tv=
240s
Den
sité
(g/c
m3 )
1.03
1.19
1.19
1.22
1.23
1.32
1.19
1.32
1.32
1.22
Dur
eté
shor
e A
(3")
4669
7075
6971
7272
7472
Rés
ista
nce
à la
rupt
ure
(MPa
)2.
921
.522
.124
.519
.713
.127
.815
.824
.921
.1A
llong
emen
t à la
rupt
ure
(%)
270
364
345
280
340
316
300
394
240
400
Mod
ule
à 50
% d
'allo
ngem
ent (
MPa
)0.
72.
12.
22.
92.
22.
52.
42.
22.
82.
4M
odul
e à
100%
d'al
long
emen
t (M
Pa)
1.2
4.6
4.9
7.4
4.8
4.9
5.4
4.2
7.7
5M
odul
e à
200%
d'al
long
emen
t (M
Pa)
-11
.913
--
10.7
178.
9-
13M
odul
e à
300%
d'al
long
emen
t (M
Pa)
-18
.419
.8-
18.3
27.8
27.8
12.7
-18
.1Tg
mid
poin
t (°C
)-2
7-3
3-2
8-2
8-2
7-2
9-2
8-2
8-2
6-1
3R
ebon
d (%
)-
3333
2528
2524
2930
11G
onfle
men
t (ta
ux d
e vu
lcan
isat
ion)
-1.
821.
82-
-1.
391.
891.
83-
1.66
Rés
ista
nce
à l'a
bras
ion
(mm
3 )81
9696
8911
915
890
153
9489
Com
pres
sion
set (
%) :
70H
à 1
20°C
-19
1923
2025
2334
2524
A
T
able
au 4
: ta
blea
u gl
obal
des
car
acté
rist
ique
s des
mat
éria
ux d
e l’é
tude
, dét
erm
inée
s à te
mpé
ratu
re a
mbi
ante

Chapitre 2 : Matériaux de l’étude et principaux moyens de caractérisation mis en œuvre
78
III.3. Méthodes de préparation des matériaux
Dans le cadre de cette étude, les matériaux sont préparés suivant deux formes : des joints
injectés ou des plaques pressés.
En effet, dans la première partie de cette analyse, nous examinerons la tenue des joints en
conditions quasi-réelles, au regard des paramètres matériaux. Ainsi, la société Paulstra a préparé
des séries de joints correspondant aux diverses séries présentées ci-dessus, dans leurs conditions
usuelles de préparation, c’est-à-dire par injection.
Ensuite, dans une seconde partie, les analyses se feront sur des appareils d’expérimentation de
laboratoire. Ainsi, pour effectuer ces tests, nous disposerons de plaques de matériaux, préparées
par pressage/moulage.
On peut raisonnablement supposer que ces préparations très proches ne devraient pas engendrer
de propriétés ou de comportements différents de nos matériaux, selon qu’ils soient injectés ou
pressés.

Chapitre 2 : Matériaux de l’étude et principaux moyens de caractérisation mis en œuvre
79
IV. Différentes techniques expérimentales utilisées
IV.1. Caractérisations tribologiques
IV.1.1 Bancs d’essai SNR
Afin de tester l’étanchéité statique et dynamique des joints de roulements de roue de véhicule de
tourisme, la société SNR Roulements dispose de 2 types de banc d’essai :
Des bancs BFRE (Banc Fusée Roue Etanchéité) : les joints sont montés sur roulement avec
les pièces constructeurs environnantes et la durée d’essai est une variable expérimentale;
Des bancs BDJE (Banc D’essai des Joints d’Etanchéité) : les joints sont montés uniquement
avec les bagues du roulement et l’essai va jusque la perte d’étanchéité du joint (la durée
d’essai est un résultat du test).
IV.1.1.1. Banc Fusée Roue Etanchéité
L’essai BFRE consiste à tester en endurance les joints d’étanchéité pour roulements de
roue dans des conditions représentatives des conditions d’exploitation de ces roulements
(montage similaire au montage constructeur et application de chargements radiaux et axiaux).
Une buse de projection alimentée par une centrale à boue projette de la boue calibrée sur la face
intérieure du roulement selon un cycle bien défini. La projection de boue s’effectue au niveau de
l’axe horizontal du roulement.
La boue utilisée dans ces essais est une boue calibrée (dénommée boue d’Arizona) toujours de
même composition et dans les mêmes proportions, pour assurer au mieux la reproductibilité des
essais. La composition de cette boue est la suivante :
∑ Sable BTF (Si, O) de granulométrie variée mais dont la taille de la majorité des
particules est comprise entre 75 et 105 µm, environ 30,8 % en poids ;
∑ Silice C300 (Si, O, Al, K) de granulométrie 15 µm, environ 30,8 % en poids ;
∑ Argile de Provin (kaolin) (Si, Al, O), environ 11,5 % en poids ;
∑ Sel gemme, environ 15,4 % en poids ;
∑ Précipités de Carbonate de Calcium et du Chlorure de Calcium soluble, environ 10 %
en poids au total.
Les essais sont réalisés à température ambiante,avec des durées variables, comprises entre 200h et
400h dans notre étude. Le système présente des chargements axiaux et radiaux variables, et une
vitesse de rotation de 1000 tr/min (environ 1m/s).

Chapitre 2 : Matériaux de l’étude et principaux moyens de caractérisation mis en œuvre
80
Le roulement servant de support pour les essais d’étanchéité du joint dont il est équipé est
placé dans un ensemble représentatif du montage constructeur (embout de transmission – moyeu
– boîtier). Cet ensemble est associé à un boîtier étanche comprenant un système de projection
(buse). Un plateau monté en lieu et place de la jante est fixé sur l’ensemble du montage. Ce
nouvel ensemble est positionné à l’extrémité d’une broche avec un centrage en rotation par
rapport à l’axe de la broche.
IV.1.1.2. Banc D’essai des Joints d’Etanchéité
Sur le banc BDJE, le joint est sollicité suivant les procédures d’essais internes à SNR. De
plus, afin de reproduire les effets soumis au joint et au déflecteur, il est possible d’intégrer un
mouvement de battements radial et axial qui simule les charges appliquées au roulement dans ses
applications courantes.
Les essais sont réalisés à température ambiante, avec une vitesse de rotation de 1000 tr/min
(correspondant environ à 1 m/s) de la bague intérieure. Concernant la présence de boue, il y a
immersion dans le polluant (constitué de boue d’Arizona, décrite précédemment) jusqu’au centre
de l’axe du joint.
En ce qui concerne le suivi de l’essai, dès que le polluant franchi le joint, il y a contact avec un fil
capteur qui déclenche le signal de présence d’eau et arrête le banc. On détermine ainsi la durée de
vie du joint testé. Puis l’analyse du système après essai s’effectue par une observation de l’usure
des lèvres et des surfaces de frottement (moulage, photo), et éventuellement des mesures des
zones usées.
IV.1.2 Tribomètre bille/plan
Ce tribomètre, conçu et développé au Laboratoire de Tribologie et de Dynamique des
Systèmes de l’Ecole Centrale de Lyon (LTDS) (cf. Figure 6), nous permet de tester le frottement
d’un indent sphérique type Bille (100Cr6) au contact de l’un de nos matériaux sous forme d’un
disque mis en rotation.
Durant cet essai, nous faisons l’acquisition de différents paramètres :
- la force normale appliquée par un système de ressorts Fn(t) ; la force tangentielle résultante
mesurée par des jauges de contraintes Ft(t) : nous obtenons ainsi la valeur du coefficient de
frottement µ(t)=Ft(t)/Fn(t) ;
- h(t) où h est l’enfoncement de la bille dans le matériau, mesuré par le biais d’une fibre
optique, placée sur le côté du porte-bille fixe ;
- la température mesurée au-dessus de la bille grâce à un thermocouple T°(t).

Chapitre 2 : Matériaux de l’étude et principaux moyens de caractérisation mis en œuvre
81
disque d’élastomère
moteur
bille 100Cr6
socle en rotationsupport
massif fixe
FFnn
FFtt
T°T°
hhporte-bille
Figure 6 : Représentation schématique du tribomètre bille/plan
Les échantillons sont découpés dans les plaques moulées à l’aide d’un emporte-pièce afin
d’obtenir des disques de diamètre 36 mm et d’une épaisseur de 2mm. Ceux-ci, ainsi que la bille en
100Cr6 servant d’indenteur, sont nettoyés avec de l’alcool et du papier absorbant.
La bille est fixée dans son porte-bille et le disque est collé sur le socle en rotation.
Concernant la force normale appliquée Fn, nous avons cherché à transposer le niveau de
contrainte en bout de lèvre à la pression maximale du contact bille/plan de notre tribomètre.
Ainsi, par l’intermédiaire d’une modélisation et d’un calcul en statique par élément fini de la lèvre
au contact avec le déflecteur, l’état de contrainte en bout de lèvre a pu être approchée. Puis, en se
basant sur la théorie du contact bille/plan définie par Hertz dans le cas d’un massif semi-infini,
une force normale de l’ordre de 5,5 N semblait alors appropriée. Cette méthode fait appel à
quelques approximations, mais fourni une valeur approchée qui nous semble tout a fait correct.
Pour simuler le comportement d’un joint d’un véhicule se déplacant à 50km/h, il nous faut alors
mettre en rotation notre système afin d’obtenir une vitesse de rotation de 400tr/min, soit pour un
diamètre de trace de 20mm, une vitesse de glissement de 420mm/s. Par simplicité et afin de
quantifier l’usure, 1 heure d’essai à température ambiante a été choisie.
En résumé, les paramètres utilisés sont :
- Fn=5,5N
- Vg=420mm/s
- Tamb.
- 24000 tours soit 1h d’essai.

Chapitre 2 : Matériaux de l’étude et principaux moyens de caractérisation mis en œuvre
82
IV.2. Caractérisations mécaniques
IV.2.1 Spectrométrie mécanique
La spectrométrie mécanique permet l’étude des propriétés viscoélastiques d’un matériau par
la mesure de son module dynamique en fonction de la température à une fréquence donnée
(mesures isochrones) ou en fonction de la fréquence de sollicitation à une température donnée
(mesures isothermes). L’échantillon est sollicité par l’application d’une contrainte σ (ou d’une
déformation ε ) sinusoïdale.
σ* = σ0.exp(iωt) ou ε* = ε0.exp(iωt)
Pour rester dans le domaine linéaire, l’amplitude de la sollicitation est maintenue faible, de l’ordre
de 10-5. La réponse en régime linéaire est également sinusoïdale et déphasée d’un angle δ par
rapport à la sollicitation.
ε* = ε0.exp(iωt-δ) ou σ* = σ0.exp(iωt-δ)
Le module dynamique E* s’exprime ainsi :
E* = σ*/ε* = E’ + iE’’
Où E’ est le module de conservation ou élastique en lien avec l’énergie élastique emmagasinée de
manière réversible dans le matériau, E’’ est le module de perte ou visqueux correspondant à
l’énergie dissipée dans le matériau.
Le déphasage entre la contrainte et la déformation donne accès au coefficient de frottement
intérieur qui caractérise l’énergie dissipée sous forme de chaleur par rapport au maximum
d’énergie élastique stockée au cours d’un cycle. Le facteur de perte s’exprime ainsi :
tan δ = E’’ / E’
Dans le cas de sollicitation en cisaillement ces définitions sont transposables et le module E
devient le module G. Les variations du module complexe et du facteur de perte tan δ, en fonction
de la température ou de la fréquence, permettent de caractériser les relaxations mécaniques
associées aux divers degrés de liberté des chaînes macromoléculaires.
IV.2.1.1. Domaine linéaire_Le pendule de torsion
Dans le cadre de notre étude, les mesures de module dynamique ont été réalisées en fonction
de la température sur des pendules de torsion inversés fonctionnant en régime harmonique. Ce
dispositif a été mis au point au sein du Groupe d’Etude de Metallurgie Physique et de Physique
des Matériaux de l’Insa de Lyon (GEMPPM).

Chapitre 2 : Matériaux de l’étude et principaux moyens de caractérisation mis en œuvre
83
a) PRINCIPE DE FONCTIONNEMENT DU PENDULE DE TORSION
axe d’équilibrage
miroir
bobines fendues
tige de suspension
Dewar (isolation thermique)
mors supérieur
fouréchantillon
mors inférieur
fil de suspension
aimant
fléau de suspension
contre poids
bâti
Figure 7 : Schéma du pendule de torsion
miroir
cellulesaimant
bobine
Faisceau lumineux
Figure 8 : Schéma du pendule ; vue de dessus
Le montage expérimental est représenté sur la Figure 7 et la Figure 8. L’échantillon est
sollicité en torsion sinusoïdale, à des taux de déformation faibles (10-5). Travailler à des taux de
déformation de cet ordre de grandeur permet de s’assurer que les échantillons sont sollicités dans
le domaine linéaire et permet d’éviter l’apparition des effets Payne et Mullins, présentés dans la
partie bibliographique, qui pourraient affecter les mesures.
La base de l’échantillon est reliée au bâti, et donc immobile. Le haut de l’échantillon est relié
à une tige sur laquelle sont fixés un miroir et un aimant. En modulant le courant dans les bobines
placées de part et d’autre de l’aimant, le champ magnétique est modifié, ce qui provoque la
rotation de l’aimant. L’échantillon est donc sollicité en torsion. Un faisceau lumineux est envoyé
sur le miroir et réfléchi vers deux cellules photosensibles différentielles qui enregistrent donc la
déformation de l’échantillon. Le haut de la tige est relié à un fil. Ainsi, si l’échantillon est sollicité à

Chapitre 2 : Matériaux de l’étude et principaux moyens de caractérisation mis en œuvre
84
des petites déformations, aucune force ne vient s’opposer à la rotation de la tige et perturber la
mesure. Le système est maintenu à l’équilibre par un contrepoids, et l’échantillon n’est donc
soumis à aucune force de pesanteur.
Un four est placé autour de l’échantillon afin d’effectuer des balayages en température pendant
les mesures. Le four est lui-même entouré par un Dewar qui assure l’isolation thermique avec
l’extérieur. Le Dewar peut recevoir de l’azote liquide, ce qui permet d’effectuer des mesures à
basses températures (jusqu’à 173K environ).
L’ensemble du système échantillon/chauffage/acquisition est contrôlé par une chaîne
d’acquisition numérique. Les taux de contraintes et déformations peuvent être asservis, ainsi que
les variations de température. Dans notre cas, les mesures sont effectuées en déformation
asservie.
b) PARAMÈTRES CARACTÉRISTIQUES DE NOS ESSAIS
Tous les échantillons sont sollicités en torsion sinusoïdale à une fréquence de 0,1 Hz
(mesures isochrones) sur une rampe de température allant de 173 K à 353 K ou 453K, suivant le
pendule utilisé, à 1 K/minute. La température de transition vitreuse Tg des échantillons se situant
entre 238 K et 260 K, cela nous permet d’obtenir un plateau vitreux suffisant pour déterminer le
module vitreux (servant de référence pour les résultats) et aussi de suivre l’évolution des
propriétés à des températures proches de celles d’utilisation en conditions réelles (≈353 K ou
même plus).
Les éprouvettes testées sont, en général, des parallélépipèdes rectangles de longueur 15 mm,
de largeur 6 mm et d’épaisseur 2 mm environ, issues de plaques d’élastomères. Dans le cas de
l’étude des lèvres réelles, elles ont été découpées au cutter selon les dimensions suivantes :
longueur 4-6 mm, largeur 0,9-1,4 mm et épaisseur 0,5 mm (environ).
Le module de cisaillement complexe est calculé de la manière suivante :
∗
∗∗
ΘΓ
⋅=f
G 1
où Γ* est le couple de torsion et Θ* est la déformation angulaire de l’échantillon. Le facteur de
forme f pour des échantillons de forme parallélépipède rectangle s’écrit :
Llef ⋅⋅
=3β où β varie en fonction de e/l.
IV.2.1.2. Domaine non-linéaire_Viscoanalyseur RDS
Les mesures pour des amplitudes plus importantes (dans le domaine non-linéaire) permettent
d’étudier l’effet Payne (cf. Chapitre 1). Ces mesures ont été réalisées sur un viscoanalyseur de type

Chapitre 2 : Matériaux de l’étude et principaux moyens de caractérisation mis en œuvre
85
METRAVIB RDS VA 2000 en cisaillement plan. Les mesures consistent à déterminer le module
complexe G*, défini précédemment, à une température et une fréquence données en fonction de
la déformation (gamme de fréquence disponible comprise entre 0,001 Hz et 200 Hz).
La technique consiste à appliquer une déformation sinusoïdale à un échantillon et à mesurer la
contrainte résultante, réponse de l’échantillon (cf. Figure 9). Aux grandes déformations, la
réponse peut devenir non-linéaire, c’est-à-dire non sinusoidale. Il faut donc être conscient que le
module est calculé à partir de la seule fondamentale du signal réponse.
εr
εr
).sin(.0 tωεε =).sin(.0 δωσσ += t
échantillon
Figure 9 : Schéma de principe du viscoanalyseur
a) PROCÉDURE EXPÉRIMENTALE
L’échantillon est un parallélépipède de dimension voisines de 15×5×2 mm3, collé à des mors
adaptés (Figure 9), et sollicité en cisaillement plan.
Concernant la gamme de déformation appliquée, un balayage en déformation dynamique [10-6m ;
5.10-4m] a été opéré. Les échantillons ayant tous une épaisseur égale à 2 mm, cela est équivalent à
un balayage en cisaillement dynamique γ compris entre 5.10-4 et 0,25.
Deux températures (30°C et 70°C) ont été choisies pour effectuer les essais. A 30°C, nous
sommes dans des conditions de température au repos du roulement ; tandis que 70°C est une
température représentative de la température d’utilisation du roulement.
b) PRÉCAUTIONS PARTICULIÈRES
Après le collage de l’échantillon sur les mors (passage très délicat de la manipulation, les
échantillons devant être bien parallèles aux mors) et le temps nécessaire pour une mise en
température de l’échantillon (environ 1h), celui-ci est sollicité une première fois de manière à
éliminer l’effet Mullins (cf. Chapitre 1).
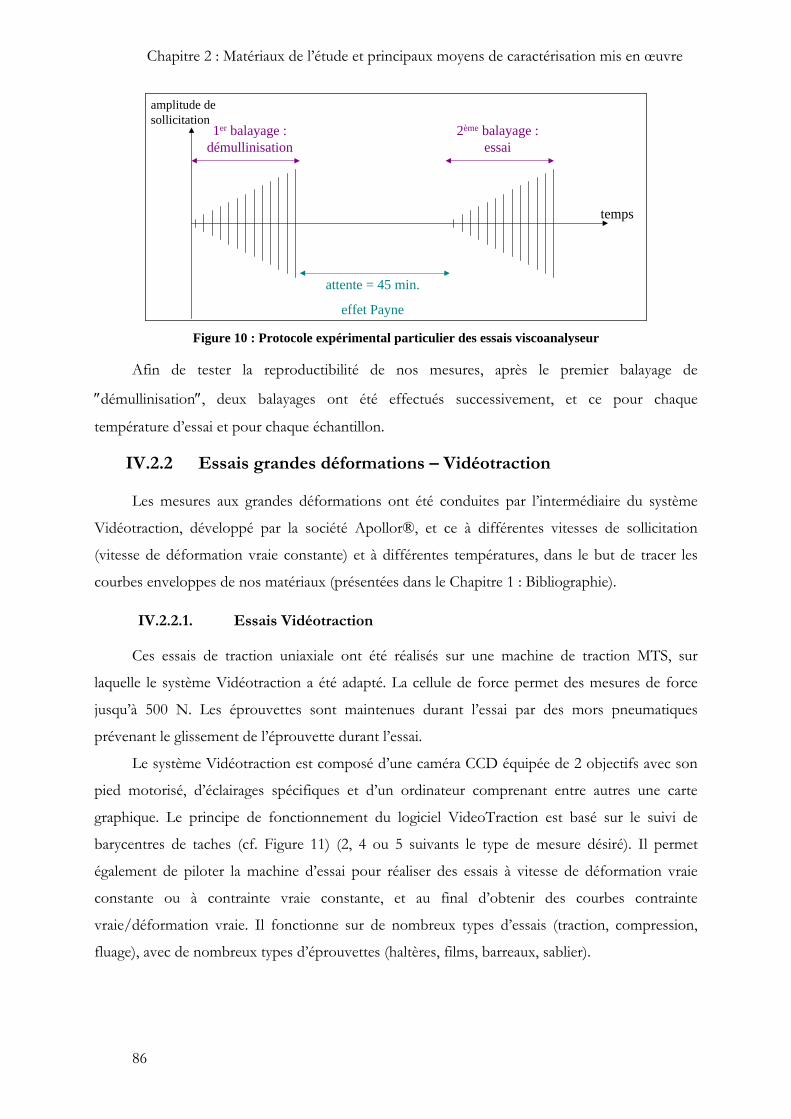
D’autre part, pour chaque échantillon, un délai d’attente de 45 minutes a été nécessaire
entre chaque essai, de manière à recouvrer la chute de module due à l’effet Payne, provoqué par
le balayage en déformation (ce délai a été déterminé dans le cadre d’une étude précédente sur les
mêmes matériaux).
Chapitre 2 : Matériaux de l’étude et principaux moyens de caractérisation mis en œuvre
86
temps
attente = 45 min.
effet Payne
1er balayage :démullinisation
2ème balayage :essai
amplitude desollicitation
Figure 10 : Protocole expérimental particulier des essais viscoanalyseur
Afin de tester la reproductibilité de nos mesures, après le premier balayage de
″démullinisation″, deux balayages ont été effectués successivement, et ce pour chaque
température d’essai et pour chaque échantillon.
IV.2.2 Essais grandes déformations – Vidéotraction
Les mesures aux grandes déformations ont été conduites par l’intermédiaire du système
Vidéotraction, développé par la société Apollor®, et ce à différentes vitesses de sollicitation
(vitesse de déformation vraie constante) et à différentes températures, dans le but de tracer les
courbes enveloppes de nos matériaux (présentées dans le Chapitre 1 : Bibliographie).
IV.2.2.1. Essais Vidéotraction
Ces essais de traction uniaxiale ont été réalisés sur une machine de traction MTS, sur
laquelle le système Vidéotraction a été adapté. La cellule de force permet des mesures de force
jusqu’à 500 N. Les éprouvettes sont maintenues durant l’essai par des mors pneumatiques
prévenant le glissement de l’éprouvette durant l’essai.
Le système Vidéotraction est composé d’une caméra CCD équipée de 2 objectifs avec son
pied motorisé, d’éclairages spécifiques et d’un ordinateur comprenant entre autres une carte
graphique. Le principe de fonctionnement du logiciel VideoTraction est basé sur le suivi de
barycentres de taches (cf. Figure 11) (2, 4 ou 5 suivants le type de mesure désiré). Il permet
également de piloter la machine d’essai pour réaliser des essais à vitesse de déformation vraie
constante ou à contrainte vraie constante, et au final d’obtenir des courbes contrainte
vraie/déformation vraie. Il fonctionne sur de nombreux types d’essais (traction, compression,
fluage), avec de nombreux types d’éprouvettes (haltères, films, barreaux, sablier).

Chapitre 2 : Matériaux de l’étude et principaux moyens de caractérisation mis en œuvre
87
a) PROTOCOLE EXPÉRIMENTAL SPÉCIFIQUE À NOS MESURES
Des éprouvettes en forme d’haltères, découpées dans les plaques moulées, offrent une longueur
utile de 20 mm pour une largeur de 4 mm et une épaisseur de l’ordre de 2 mm. 4 taches sont
disposées, comme représenté sur la Figure 11, de manière à mesurer la déformation dans la
direction de traction (ε1) et celle dans la direction perpendiculaire (ε2).
Figure 11 : Eprouvette + taches pour VideoTraction
Ces mesures permettent de suivre d’une part la déformation vraie (ε1) mais aussi la contrainte
vraie définie par :
σvraie=F/Svraie avec Svraie= S0.(1+ε2)²
On peut ainsi pour chaque essai tracer la courbe σvraie=f(εvraie).
Nos différents tests ont été réalisés à vitesse de déformation vraie constante, afin de s’affranchir
de la dépendance des propriétés des polymères avec la vitesse de sollicitation.
IV.2.2.2. Propriétés à la rupture - Courbes enveloppes
Dans le but de tracer les courbes enveloppes de nos matériaux, les tests ont été réalisés à
différentes températures (T° = 30°C, 70°C et 110°C) et à différentes vitesses de déformation
vraie ( ε°vraie= 2.10-2 ; 5.10-3 ; 10-3).
Afin d’augmenter la précision de nos mesures de contrainte vraie à la rupture σr et déformation
vraie à la rupture εr, pour chaque couple vitesse/température, 5 ou 6 éprouvettes d’un même
matériau ont été testées.
IV.2.3 La Nanoindentation
Introduites au début des années 1980 ([Pethica, Hutchings et al., 1983] ; [Loubet, Georges et
al., 1984]), les mesures de nanoindentation instrumentée se sont aujourd’hui beaucoup
développées. Cette technique permet de caractériser mécaniquement les surfaces des matériaux
avec des profondeurs de l’ordre de quelques dixièmes à quelques dizaines de micromètres. Le
principe consiste en l’application d’une charge, par l’intermédiaire d’un indenteur, sur une surface.
12

Chapitre 2 : Matériaux de l’étude et principaux moyens de caractérisation mis en œuvre
88
L’appareil mesure l’enfoncement ainsi que des grandeurs (raideur, phase…) correspondant à la
réponse du matériau vis-à-vis de la sollicitation. Parmi les propriétés mesurables, on peut citer :
modules d’Young et de Coulomb, coefficient de Poisson, dureté, viscosité, forces d’adhésion
(entre la pointe et le substrat) ou encore ténacité et énergie de rupture.
IV.2.3.1. Dispositif expérimental
Nos essais de nanoindentation sont réalisés avec un Nanoindenter II, commercialisé par
MTS® et dont un schéma de fonctionnement est représenté sur la Figure 12.
Figure 12 : Schéma de principe du nanoindenteur
L’application de la charge P est réalisée par l’intermédiaire d’un solénoïde (C). La force
électromagnétique induite par le passage d’un courant à l’intérieur de la bobine génère une force
dans l’axe de la colonne, qui se répercute au niveau de l’indenteur (B) sur la surface de
l’échantillon (A). Les ressorts de rappel et de maintien (D) assurent le guidage de la colonne. Ils
maintiennent la colonne perpendiculaire à la surface des échantillons. Leur raideur est de
100N/m. La mesure du déplacement de la colonne est réalisée par un capteur capacitif (E).
L’ensemble est monté sur un bâti (F) d’une très grande rigidité (sa raideur est de 7.106 N/m). Le
porte échantillon (A) permet de disposer plusieurs échantillons dans le dispositif.
Chaque échantillon est collé sur un plot cylindrique de 32 mm de diamètre. Un objectif de
microscope permet de repérer l’endroit sur l’échantillon où vont être réalisés les essais, ainsi que
de visualiser les indents après les essais. Une table à mouvements croisés permet le
positionnement des échantillons sous la tête de mesure avec une précision supérieure au
micromètre. L’amplitude en force de la tête de mesure utilisée est de 500 mN et sa résolution de
10 nN ; son amplitude en déplacement est de 100 µm et sa résolution de 0,5 nm.
Les indenteurs en diamant utilisés pour notre étude ont une géométrie tétraédrique de type
Berkovich. L’indenteur Berkovich possède un angle entre les arêtes de 115,12° ; sa géométrie est

Chapitre 2 : Matériaux de l’étude et principaux moyens de caractérisation mis en œuvre
89
équivalente à un cône de demi-angle au sommet de 70,32° pour l’aire de contact [Johnson, 1994].
Cette transformation permet d’appliquer les formules simples de la théorie du contact ; bien que
celles-ci engendrent des approximations, nous pouvons considérer les valeurs déterminées
comme suffisamment bien approchées. D’autres types d’indenteurs à géométrie pyramidale ou
sphérique peuvent également être utilisés, ils permettent d’avoir accès à d’autres niveaux de
déformation et vitesses de déformation.
IV.2.3.2. Nos essais de Nanoindentation
Dans le cadre de notre étude, deux séries d’essais de nanoindentation ont été réalisées :
∑ d’une part, sur les lèvres réelles testées au préalable sur BFRE ;
∑ d’autre part, sur des plaques de matériaux, usées au tribomètre bille/plan ; une zone de la
partie neuve et une zone de la trace d’usure étant caractérisées.
Dans chaque cas, une préparation des échantillons ainsi qu’une procédure expérimentale
particulières ont été utilisées.
a) PRÉPARATIONS DES ÉCHANTILLONS
Dans le cadre des mesures sur les joints testés sur BFRE, les échantillons testés ont été au
préalable montés dans de la résine (polyuréthane) dans les laboratoires de SNR. La résine est
supposée être inerte chimiquement vis à vis de nos élastomères. Ensuite, une coupe est réalisée et
la surface à analyser est polie. Un point important à noter est que le polissage (de l’ordre du
micron) doit être très méticuleux afin que la surface d’essais soit parfaitement plane. Et ceci pour
deux raisons : pour éviter tout endommagement de la pointe très fragile de l’indenteur, et pour
que la rugosité soit bien inférieure à la profondeur d’indentation.
Pour ce qui est des essais sur plaques, les disques ont tout d’abord été usés sur le
tribomètre bille/plan, puis une portion de chaque disque a été découpée et collée sur leur plot.
Ainsi, nous avons pu caractériser le matériau neuf en effectuant les essais en dehors de la trace
d’usure, et le matériau usé en effectuant les essais dans la trace d’usure. Le problème de la
rugosité des échantillons évoqué précédemment est, bien entendu, tout aussi crucial dans ce type
d’essai. Malheureusement, dans le cas de ces échantillons, il est dépendant de la mise en œuvre
des plaques, et son influence sera examinée ultérieurement.

Chapitre 2 : Matériaux de l’étude et principaux moyens de caractérisation mis en œuvre
90
b) PROCÉDURES EXPÉRIMENTALES
(i) Essais sur lèvres injectées – méthode quasi-statique :
L’indenteur est tout d’abord chargé puis déchargé trois fois, de manière consécutive, avec
une vitesse de chargement constante (Figure 13). Chaque déchargement est stoppé lorsque la
charge atteint 10% de la valeur maximale obtenue, afin d’être certain que le contact entre la
pointe et l’échantillon soit maintenu. Ces cycles sont effectués pour tenir compte de la réponse
viscoélastique des matériaux. Une fois ces trois cycles effectués, la charge est maintenue à un
plateau de 10% de la charge maximale pendant une durée de 100s, afin de prendre en compte les
dérives thermiques du système. En effet, même si tout le système est contrôlé en température à
±1°C, de petites dilatations parasites de l’appareillage peuvent apparaître et modifier les mesures.
Enfin, une autre période de maintien est réalisée à charge constante maximale afin de minimiser
les problèmes de fluage liés au caractère viscoélastique de nos matériaux. Effectivement, en
l’absence de ce segment de maintien à charge maximale, l’enfoncement continue d’augmenter en
début de décharge et ne permet plus un calcul correct du module([Hochstetter, Jiminez et al.,
1999]). On a vérifié, comme le montre la Figure 13 (droite), qu’un temps de maintien de 100s
permettait de s’affranchir en grande partie de ce problème.
0
2
4
6
8
10
12
0 50 100 150 200 250 300temps (s)
forc
e (m
N)
1000
1050
1100
140 170 200 230 260temps (s)
dép
lace
men
t (n
m)
Figure 13 : Procédure d’indentation ; profil de charge tous segment(gauche) ; zoom sur le maintien à Fmax,
phénomène de fluage à charge constante dans les polymères (droite)
La Figure 14 représente une courbe typique d’indentation d’un matériau élastoplastique (de
type silice) : une première charge suivie par une décharge. Même si dans l’étude des élastomères
en nanoindentation, on peut noter certaines spécificités, détaillées par la suite, cette courbe
‘modèle’ permet d’appréhender la logique de calcul des propriétés mécaniques des échantillons à
partir d’un essai de nanoindentation.
Lors de la décharge, le matériau a un comportement élastique, ce qui permet d’obtenir les
relations entre la courbe de décharge et le module élastique à partir de la théorie de l’élasticité. On
note hmax la profondeur maximale atteinte par l’indenteur dans le matériau. Cette profondeur est
obtenue à la charge maximale Pmax. On note hr la profondeur résiduelle de l’indent après que

Chapitre 2 : Matériaux de l’étude et principaux moyens de caractérisation mis en œuvre
91
l’indenteur ait été enlevé. Dans le cas d’un élastomère, le retour est totalement élastique, et le
matériau ne présente donc pas d’empreinte résiduelle ; cependant, ce retour prends du temps du
fait du caractère viscoélastique des matériaux. L’intersection entre l’axe des abscisses et la
tangente à la courbe de décharge au point de charge maximum est notée hr’.
Figure 14 : Exploitation des courbes d’indentation
La pente S de la tangente à la courbe de décharge au point de charge maximum représente la
raideur de contact en ce point. Directement mesurée par le nanoindenteur, la raideur de contact
est à la base des calculs des principales propriétés mécaniques du matériau indenté ; on a :
maxmax
' maxmaxh
h
r SPh
hPPhh ⎟
⎠⎞
⎜⎝⎛−=⎟
⎟
⎠
⎞
⎜⎜
⎝
⎛
∂∂
−=
Pour déterminer le module d’élasticité équivalent E*, défini par 122 11
−
∗⎟⎟⎠
⎞⎜⎜⎝
⎛ −+
−=
i
i
m
m
EEE νν (Em et Ei
sont respectivement les modules d’Young du matériau et de l’indenteur, et νm et νi les coefficients
de Poisson du matériau et de l’indenteur), à partir d’un essai avec une pointe axysymétrique,
[Loubet, Georges et al., 1984] utilise l’équation suivante, démontrée par [Sneddon, 1965] :
∗∗ ⋅⋅=⋅⋅=⎟⎠⎞
⎜⎝⎛
∂∂
= EAaEhPS N
hh π
22max
max
où a est le rayon de contact et AN la projection de l’aire de contact, calculé à partir de la fonction
de forme de l’indenteur AN=f(hr’). Cette dernière est approximée par une fonction polynomiale
de degré 6, et est déterminée par une calibration au préalable sur du verre.
N.B. : Les indenteurs coniques, tétraédriques ou pyramidaux ne sont jamais parfaitement pointus : la pointe est
plus ou moins émoussée car l’usinage n’est jamais parfait. Il est possible d’estimer le défaut de pointe en suivant le
protocole établit par [Loubet, Bauer et al., 1993] sur un matériau de référence tel que la silice fondue. Cependant,
pour de nombreux auteurs ([Briscoe, Fiori et al., 1998], [Hochstetter, Jiminez et al., 1999]…), l’utilisation d’un
tel substrat rigide introduit une estimation incorrecte du défaut de pointe pour une utilisation avec des polymères.

Chapitre 2 : Matériaux de l’étude et principaux moyens de caractérisation mis en œuvre
92
(ii) Essais sur plaques moulées – méthode dynamique :
Le premier point particulier relatif à cette étude concerne la vitesse de sollicitation. En
effet, les propriétés mécaniques des polymères dépendent de la fréquence à laquelle elles sont
mesurées. Il apparaît donc important de réaliser les essais sur des polymères à vitesse de
déformation constante. Une procédure a donc été développée par [Lucas, Oliver et al., 1997] afin
de maintenir hh••
=ε constant, ce qui se traduit par maintenir PP•
constant [Briscoe, Fiori et al.,
1998] (pour les matériaux présentant une dureté constante avec l’enfoncement) ; ce qui revient à
charger et à décharger de façon exponentielle avec le temps.
La deuxième particularité de ces essais est l’utilisation du mode dynamique (« Continuous
Stiffness Measurement »). Cette méthode mise au point par [Loubet, Bauer et al., 1993] et
[Pethica, Hutchings et al., 1983] permet de s’affranchir de deux inconvénients majeurs de la
méthode quasi-statique présentée précédemment :
∧ Lorsque l’on cherche à déterminer les propriétés d’un matériau à plusieurs profondeurs,
c’est autant d’essais supplémentaires qu’il faut répéter ;
∧ Alors que la décharge a déjà commencé, l’enfoncement continue d’augmenter, du fait du
caractère viscoélastique des matériaux étudiés ; ainsi, cette forme arrondie en début de décharge
ne permet plus de calculer la raideur de contact S correctement.
Le principe de la méthode dynamique repose sur la superposition d’un mouvement continu
(chargement exponentiel) et d’un mouvement oscillant à fréquence fixe et de faible amplitude
(quelques nm). La fréquence utilisée pour nos essais sera de 32 Hz.
Cette méthode permet donc de caractériser le comportement viscoélastique du matériau à tout
moment au cours de l’essai, à une fréquence donnée. En effet, dans le cas d’un poincçon
axisymétrique, la déformation relative moyenne est considérée constante au cours d’un essai
d’indentation (de l’ordre de 6,9% pour un indenteur Berkovitch, par exemple).
Dans le cas des matériaux de l’étude, le module de restitution G’ et la tangente de l’angle de perte
tan δ sont calculés dans la phase de chargement. Le module de restitution est donné par une
relation liant la raideur de contact S mesurée par la machine et le rayon de contact ; une formule
analogue détermine le module de dissipation G’’, en rapport avec l’amortissement mesuré par la
machine ; on peut ainsi en déduire la valeur de la tangente de l’angle de perte :
haSG ∝′ et haCG ω∝′′ et SCGG ωδ =′′′=tan

Chapitre 2 : Matériaux de l’étude et principaux moyens de caractérisation mis en œuvre
93
IV.3. Caractérisations physico-chimiques
IV.3.1 Analyse ThermoGravimétrique (ATG)
La thermogravimétrie est une technique permettant de mesurer de très faibles évolutions de
masse sous l’action d'une variation de température. L’utilisation de différentes vitesses
d’évolution de la température permet d’observer les cinétiques de dégradation propres à chacun
des constituants. L’observation de ces pertes de masses peut se faire sous différentes atmosphères
: azote, air.
Le protocole utilisé est largement inspiré de la littérature concernant la caractérisation des
élastomères chargés. La masse de l’échantillon est de l’ordre de 15 mg. Une isotherme à 35°C
pendant 1 heure permet de placer chaque échantillon dans les mêmes conditions de départ. On
effectue ensuite une montée en température à une vitesse de 10°C/min de 35°C à 950°C sous
Air. Afin d’affiner les résultats dans une gamme de température plus basse, et donc plus proche
de l’utilisation réelle, un nouveau protocole a été établi : une isotherme à 35°C pendant 10
minutes, puis une montée à 1°C/min jusque 350°C, sous air. Les échantillons usés ont également
été testés avec ce second protocole.
IV.3.2 Analyse Thermique Différentielle (ATD)
Cette technique (communément nommée DSC) permet de mesurer à pression constante
Patm la capacité calorifique massique Cp en fonction de la température. Ces mesures consistent à
mesurer la différence de flux de chaleur entre 2 coupelles, l’une vide servant de référence, et
l’autre contenant l’échantillon analysé, maintenues à la même température, lors de montées
linéaires en température. La dérivée de cette énergie par rapport à la température permet de
calculer la capacité calorifique de l'échantillon. Le passage de la transition vitreuse de l’ensemble
de nos matériaux nécessite un apport d'énergie supplémentaire par rapport à la capsule de
référence. Ce passage s'accompagne donc d'une variation ∆Cp(J/g.K-1) de la capacité calorifique.
Les essais ont été réalisés sur un dispositif DSC Pyris Diamond de Perkin Elmer sur des
échantillons dont la masse est de l'ordre de 15 mg. La vitesse de balayage est égale à 10°C.min-1.
IV.3.3 Mesure des énergies de surface – Méthode de la goutte posée
La mesure de l’énergie de surface de solides s’avère assez délicate et suscite de nombreuses
controverses car elle ne peut se faire directement. Une des méthodes les plus employées consiste
à déposer sur une surface plane du solide considéré, une goutte de liquide servant de sonde et
ayant des propriétés de surface connue. Cette goutte atteint une position d’équilibre et forme,

Chapitre 2 : Matériaux de l’étude et principaux moyens de caractérisation mis en œuvre
94
dans certaines conditions, un angle de contact avec le plan (cf. Figure 15). Cet angle est une
caractéristique de la différence d’énergie de surface entre le liquide et le solide. L’énergie de
surface γS qui pourra être calculé à partir de cet angle de contact dépend fortement du choix des
liquides servant de sonde ainsi que de la méthode d’exploitation des mesures.
Le point de départ de l’ensemble de ces méthodes est la relation établie par [Young, 1905], basée
sur l’angle entre le plan tangent de la goutte au point triple et la surface du solide.
Figure 15 : Angle de contact θ d’une goutte de liquide déposée sur une surface solide
La relation proposée par Young en 1805 est basée sur une analyse mécanique du phénomène :
θγγγ cosLVSLSV =− Équation 1
En combinant cette équation avec la relation établie par [Dupré, 1869] pour définir le travail
d’adhésion à des interfaces solide-liquide, on obtient la base des différentes méthodes de calcul de
l’énergie de surface à partir des mesures d’angle de contact :
SLLSLSLW γγγθγ −+=+= )cos1( Équation 2
où γS et γL sont les énergies de surface du solide et du liquide et γSL la tension interfaciale.
IV.3.3.1. Mesure des angles de contact
Pour effectuer ces mesures, nous avons utilisé le banc de mesure « Contact Angle
Goniometer » de Ramé Hart Inc., représenté sur la Figure 16, du Laboratoire des Matériaux
Macromoléculaires de l’INSA de Lyon (LMM).
Figure 16 : Schéma de principe du Goniomètre automatique : 1 - banc de mesure ; 2 – source de lumière ;
3 – porte échantillon ; 4 – porte seringue ; 5 – seringue ; 6 – caméra ; 7 et 8 – micro-ordinateur équipé
d’un analyseur d’image ; 9 –échantillon ; 10 - goutte

Chapitre 2 : Matériaux de l’étude et principaux moyens de caractérisation mis en œuvre
95
Sur la surface préalablement nettoyée (banc à ultrasons avec éthanol), nous formons, à
l’aide d’une micro seringue, une goutte de liquide sonde à l’extrémité d ‘une aiguille non
biseautée. Le profil de cette goutte déposée sur la surface à étudier, est alors observé. Il est
important de bien gérer la façon dont est déposée la goutte : après formation, la goutte doit être
amenée délicatement au contact de la surface et se détacher facilement ; on évite ainsi les effets
dynamiques d’écrasement de la goutte. Avec ce mode opératoire, la valeur de l’angle est maximale
et la reproductibilité très grande.
Dans le cadre de notre étude, les liquides sonde utilisés sont les suivants :
- le diiodométhane, comme liquide dispersif,
- l’eau, capable de former des liaisons hydrogène,
- le formamide, qui est le seul liquide à avoir une composante polaire supérieure à sa
composante hydrogène.
IV.3.3.2. Calcul de l’énergie de surface
Les méthodes de calcul de l’énergie de surface d’un matériau sont basées sur l’approche
développée par [Fowkes, 1964] en 1964. Cette approche part de l’hypothèse selon laquelle
l’énergie de surface peut-être considérée comme étant la somme des différents types
d’interactions pouvant agir à la surface d’un matériau. Ces interactions peuvent être dues aux
forces de dispersion, polaire, hydrogène, acide-base…
La méthode de calcul de [Owens et Wendt, 1969] est basée sur la résolution de l’Equation 1 par la
mesure d’angles de contact de 2 liquides (dont les caractéristiques superficielles sont connues),
l’un ayant un caractère plutôt dispersif et l’autre plutôt non-dispersif. On peut ainsi déterminer,
par la résolution du système à 2 équations, les composantes dispersive et non dispersive de la
tension superficielle du matériau.
nDL
nDS
DL
DSL γγγγθγ 22)cos1( +=+ Équation 3
Le principal reproche émis à l’encontre de cette méthode souvent utilisée est qu’elle ne permet
pas de calculer, de manière extrêmement fiable, des énergies de surface, du fait de problèmes
expérimentaux inévitables, tels que la rugosité de la surface analysée, la présence de poussière ou
de tout autre agent contaminant…Ces parasites ne sont pas pris en compte dans la théorie, et
influencent de facon notable les mesures.

Chapitre 2 : Matériaux de l’étude et principaux moyens de caractérisation mis en œuvre
96
IV.4. Caractérisations microstructurales
IV.4.1 La Microscopie
La microscopie optique et la Microscopie Electronique à Balayage (MEB) ont été utilisées
dans le cadre de l’étude des joints usés. Ainsi, l’observation de l’évolution de l’état de surface et
du profil des lèvres et des déflecteurs a pu être réalisée, et les différents modes d’usure identifiés.
La grande profondeur de champ du MEB permet une vision très complète des surfaces. Dans la
majorité des cas, il a été nécessaire de métalliser les lèvres en élastomères, afin d’évacuer au mieux
les charges créées par les électrons incidents.
La Microscopie Electronique à Balayage Environnementale (ESEM) a également été
utilisée dans le but d’observer les charges de Noir de Carbone présentes, dans les lèvres et dans
les plaques d’élastomères, notamment à cœur et en proche surface. Dans le cas de l’utilisation
ESEM, il n’est pas nécessaire de métalliser les élastomères.
Afin de compléter ces résultats et d’étudier la dispersion des charges au sein des matériaux, des
lames minces ont été préparées par Ultramicrotomie à Froid, et nous avons ainsi pu réaliser des
observations au Microscope Electronique en Transmission (MET).
IV.4.2 ESCA (Electron Spectroscopy for Chemical Analysis)
L’analyse ESCA (ou Spectrométrie de Photélectrons X, XPS) permet de différencier les
espèces et les liaisons chimiques présentes à la surface du matériau, grâce aux variations des
énergies de liaison des atomes photo-ionisés en fonction de leurs environnements chimiques
initiaux.
Il est important de noter que l’analyse n’intègre qu’une profondeur de 5 à 10nm. La surface
analysée est de l’ordre de 1200x300 µm².
IV.4.2.1. Préparation des échantillons
Différentes zones des échantillons ont été prélevées pour faire cette analyse ESCA : le cœur
du matériau, la surface neuve et la trace d’usure en surface des différents élastomères.
Figure 17 : Zones analysées des échantillons

Chapitre 2 : Matériaux de l’étude et principaux moyens de caractérisation mis en œuvre
97
Ces zones sont prélevées de l’échantillon puis placées sur un porte-échantillon. Une plaque de
cuivre perforée est ensuite plaquée sur ceux-ci laissant une zone pour l’analyse et l’autre partie
sert à évacuer les charges (dues au balayage par le canon à électrons) pour ne pas accumuler les
charges en surface et fausser l’analyse.
IV.4.2.2. Quantification des espèces chimiques
A la fin de l’acquisition d’un essais ESCA, nous obtenons un spectre sur lequel est
représenté le nombre d’électrons récoltés en fonction de l’énergie de liaison (à chaque énergie
correspond un type d’atome ou un type de liaison, suivant l’analyse) (cf. Figure 18). L’aire d’un
pic est proportionnelle au nombre d’atomes de l’élèment étudié. En calculant la part respective de
chacune de ces aires, on obtient la composition atomique de l’échantillon (sachant que ceci doit
correspondre à un bilan de 100% sur les espèces sélectionnées pour l’analyse).
Figure 18 : Exemple de spectre obtenu par analyse ESCA
De plus, la possibilité est offerte de déconvoluer le pic du Carbone en ses différentes
composantes : le Carbone graphitique (représentant les charges de Noir de Carbone) et les
liaisons macromoléculaires (C-C, C-O, C-H, C-N), principalement.

Chapitre 2 : Matériaux de l’étude et principaux moyens de caractérisation mis en œuvre
98
Références Bibliographiques
Briscoe, B. J., Fiori, L. et Pelillo, E. "Nano-indentation of polymeric surfaces." Journal of Physics - Part D : Applied Physics, 1998, vol. 31, pp. 2395-2405.
Dupré, A. "Théorie mécanique de la chaleur." Paris: Gauthier-Villars Paris, 1869, p.
Fowkes, F. M. "Attractive Forces at Interfaces." Ind. Eng. Chem., 1964, vol. 12, pp. 40-52.
Hochstetter, G., Jiminez, A. et Loubet, J.-L. "Strain-rate effects on hardness of glassy polymers in the nanoscale range. Comparison between quasi-static and continuous measurements." Journal of Macromolecular Science - Physics, 1999, vol. B38 (5&6), pp. 681-692.
Johnson, K. L. "Contact mechanics." Cambridge, G.B.: Cambridge University Press, 1994, p.
Loubet, J. L., Bauer, M., Tonck, A., et al. "Nanoindentation with a surface force apparatus".In: Mechanical properties and deformation of materials having ultra-fine microstructures. K. A. Press,1993, pp.
Loubet, J. L., Georges, J. M., Marchesini, O., et al. "Vickers indentation Curves of Magnesium Oxide (MgO)." Journal of Tribology, 1984, vol. 106, pp. 43-48.
Lucas, B. N., Oliver, W. C., Pharr, G. M., et al. "Time dependent deformation during indentation testing." Materials Research Society Symposia Proceedings, 1997, vol. 436, pp. 233-238.
Owens, D. K. et Wendt, R. C. "Estimation of the surface free energy of polymers." Journal of Applied Polymer Science, 1969, vol. 13, pp. 1741-1747.
Pethica, J. B., Hutchings, R. et Oliver, W. C. Philosophical Magazine, 1983, vol. A48 (4), pp. 593-606.
Sneddon, I. N. "The relation between load and penetration in the axysymetrique boussinesq problem for a punch of arbitrary profile." International Journal of Engineering Science, 1965, vol. 3, pp. 45-57.
Young, T. "An essay on the Cohesion of Fluids." Philoso. Trans. Royal Soc. of London, 1905, vol. 95, pp. 65-87.


















