
€¦ · · 2014-07-032 Pour tout projet de formation : +212 (0)6 61 13 69 28 ou...
69
www.cyberconseil.com www.2ssante.com CVO-CyberConseil Le partenaire des Industries de la Santé Validation / Qualification Qualité / Réglementation Gestion des risques Audits / Inspections Ingénierie / Projets Métrologie INDUSTRIES DE LA SANTÉ FORMATIONS 2010
Transcript of €¦ · · 2014-07-032 Pour tout projet de formation : +212 (0)6 61 13 69 28 ou...

www.cyberconseil.comwww.2ssante.com
CVO-CyberConseilLe partenaire des Industries de la Santé
Validation / Qualification
Qualité / Réglementation
Gestion des risques
Audits / Inspections
Ingénierie / Projets
Métrologie
INDUSTRIES DE LA SANTÉ
FORMATIONS
2010

2 Pour tout projet de formation : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
Des formateurs experts et terrains
Chaque formateur est un expert du domaine qu’il anime avec une réelle expérience terrain de plusieurs années.
Des formations pragmatiques
Nos formations contiennent des études de cas concrets basés sur des exemples rencontrés dans vos entreprises.
Une veille réglementaire quotidienne
Notre système de veille réglementaire nous permet de garantir la mise à jour de tous nos supports de formations à chaque évolution de la réglementation.
En phase avec l’évolution de vos métiers
Nos formateurs connaissent vos métiers et leur veille technologique permet d’intégrer l’état de l’art dans vos formations.
Totale indépendance
Notre centre de formation est totalement indépendant de tout prestataire, ce qui vous assure une partialité dans l’approche des différentes problématiques.
Dédié à un seul métier : la Santé
Le centre de formation CVO-CyberConseil intervient uniquement dans le secteur des Industries de la Santé pour lequel il s’est spécialisé.
Support de formation détaillé et complet
Nos supports de formation sont suffisamment détaillés et complets pour pouvoir servir de référence une fois le stage terminé.
Nombre de stagiaires limité
Nos stages ne sont pas surchargés afin de garantir un accompagnement personnalisé pendant toute la session de formation et permettre un réel échange entre les participants.
Contrôle des connaissances
En plus du contrôle de la compréhension du sujet traité par des exercices et des études de cas, chaque formation se termine par une évaluation des connais-sances réalisée avec un questionnaire à choix multiples (QCM). Pour les formations intra, une évaluation à froid est mise en place à la demande du client.
Qualité de l’accueil
Les formations se dérouleront soit en inter où les pauses café sont offertes, soit en intra.
Editorial
10 RAISONS DE CHOISIR LES FORMATIONS CVO-CYBERCONSEIL

3Pour toute formation intra-entreprise : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
B10 ICH Q10 : Mise en œuvre pragmatique
B36 APR - Annual Product Review / Revue Qualité Produit
B39 Les bonnes pratiques dans les laboratoires de chimie
C58 Mettre en place une méthode de Risk Management dans votre laboratoire pharmaceutique
C59 Gestion des risques informatiques BCP/DRP
C60 La protection contre la malveillance et l’espionnage industriel des laboratoires pharmaceutiques
C61 Gestion de la sécurité des systèmes d’informations
B104 Audit Qualité Interne et Audits Fournisseurs
B35 Les Inspections Japonaises, le MHLW et le PMDA
B34 GAMP 5 : Utilisation pragmatique dans la mise en conformité réglementaire
D12 L’Informatique : initiation pour les non spécialistes
D13 L’Automatique : initiation pour les non spécialistes
D26 De la Gestion Électronique de Documents (GED) à la Gestion Electronique de l’Information (GEI)
Depuis la création du centre de formation, nous apportons aux Industries de la Santé des formations de qualité toujours plus performantes, pragmatiques et opérationnelles, orientées résultats.
Dans notre catalogue de formations 2010, nous vous proposons de nouveaux thèmes : Gestion des risques, Métrologie, Audits / Inspections, ... et de nouvelles formations mises en place par notre équipe de formateurs experts et terrains.
Notre valeur ajoutée : nos formations sont toujours élaborées à partir de la connaissance et des problématiques liées à vos métiers. Nos intervenants vous apportent leur expérience des Industries de la Santé ainsi que des informations à jour grâce à la veille technologique et réglementaire du goupe CVO-CyberConseil.
Pour accompagner vos projets de formation orientés vers l’international, nos formations sont réalisables en anglais sur votre site ou dans notre centre de formation. Grâce à notre partenariat avec 2S Santé, nos formations peuvent aussi être réalisées au Maroc.
La liste des formations de notre catalogue n’est pas exhaustive. N’hésitez pas à nous contacter pour un besoin spécifique. Nous sommes à votre écoute pour étudier vos demandes, vous guider dans vos choix. Nous répondons à vos questions pour trouver avec vous la formation qui correspond à vos besoins.
Soy EVRARDResponsable du Centre de Formation CVO-CyberConseil
Editorial
Nouvelles formations 2010
Afin de répondre aux nombreuses demandes spécifiques de nos clients, nous vous proposons de nouvelles formations :
FORMATIONS ACTUALISÉES :Pour maintenir au plus haut niveau l’efficacité et la qualité de nos formations, nous les actualisons régulièrement.
N’hésitez pas à nous contacter. Nous sommes à votre disposition pour vous proposer des formations adaptées à vos besoins.

4 Pour tout projet de formation : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
Sen
sib
ilisa
tio
n à
la v
alid
atio
n
Valid
atio
n d
e sy
stèm
es a
uto
mat
isés
& é
qu
ipem
ents
de
pro
du
ctio
n
Valid
atio
n d
es s
ystè
mes
d’in
form
atio
n
Valid
atio
n d
es m
éth
od
es d
’an
ayse
Valid
atio
n d
es p
rocé
dés
de
net
toya
ge
Qu
alifi
cati
on
d’in
fras
tru
ctu
re In
form
atiq
ue
(Q2I
)
Valid
atio
n d
es s
ystè
mes
et
des
éq
uip
emen
ts d
e la
bo
rato
ire
(VSE
L)
ICH
Q10
: M
ise
en œ
uvr
e p
rag
mat
iqu
e
BPF
et
cGM
P : P
rin
cip
es g
énér
aux,
reto
urs
d’e
xpér
ien
ce e
t n
ou
veau
tés
Form
atio
n a
ctio
n d
es o
pér
ateu
rs s
ur s
ite
aux
BFP
et
GM
P
BPF
Co
smét
iqu
es (I
SO 2
2176
) : P
rin
cip
es e
t o
bje
ctifs
Bo
nn
es p
rati
qu
es d
e la
bo
rato
ire
po
ur l
es e
ssai
s p
récl
iniq
ues
: C
on
form
ité,
AQ
et
Au
dit
Bo
nn
es p
rati
qu
es c
liniq
ues
po
ur l
es e
ssai
s d
e m
édic
amen
t : E
thiq
ue
et q
ual
ité
des
do
nn
ées
Enre
gis
trem
ents
Ele
ctro
niq
ues
et
Ges
tio
n d
es S
ystè
mes
Info
rmat
isés
, il y
a u
n c
on
sen
sus
!
Rég
lem
enta
tio
n d
es d
isp
osi
tifs
méd
icau
x : U
SA F
DA
, Eu
rop
e M
DD
et
mar
qu
age
CE
Rég
lem
enta
tio
n a
pp
licab
le a
ux
pri
nci
pes
act
ifs A
PI /
ICH
Q7
Ass
ura
nce
Qu
alit
é et
Co
ntr
ôle
Qu
alit
é d
ans
les
lab
ora
toir
es d
e co
ntr
ôle
APR
- A
nn
ual
Pro
du
ct R
evie
w /
Rev
ue
Qu
alit
é Pr
od
uit
Les
bo
nn
es p
rati
qu
es d
ans
les
lab
ora
toir
es d
e ch
imie
An
alys
e d
e ri
squ
es C
VO
-RM
(Ris
k M
anag
emen
t)
La g
esti
on
des
ris
qu
es a
pp
liqu
ée a
ux
Dis
po
siti
fs M
édic
aux
(ISO
149
71:2
007)
La g
esti
on
des
ris
qu
es d
ans
le d
om
ain
e d
e la
san
té (I
CH
Q9)
OO
S/C
APA
– M
aîtr
iser
vo
s H
ors
Sp
écifi
cati
on
s et
op
tim
iser
vo
s A
ctio
ns
Co
rrec
tive
s
Met
tre
en p
lace
un
e m
éth
od
e d
e R
isk
Man
agem
ent
dan
s vo
tre
lab
ora
toir
e p
har
mac
euti
qu
e
Ges
tio
n d
es r
isq
ues
info
rmat
iqu
es B
CP/
DRP
La p
rote
ctio
n c
on
tre
la m
alve
illan
ce e
t l’e
spio
nn
age
ind
ust
riel
des
lab
ora
toir
es p
har
mac
euti
qu
es
Ges
tio
n d
e la
séc
uri
té d
es s
ystè
mes
d’in
form
atio
n
Au
dit
des
fou
rnis
seu
rs d
e sy
stèm
es a
uto
mat
isés
et
info
rmat
isés
Au
dit
Qu
alit
é In
tern
e - L
’ou
til d
’am
élio
rati
on
et
de
mis
e en
co
nfo
rmit
é ré
gle
men
tair
e (IS
O 1
9011
)
Au
dit
Qu
alit
é In
tern
e et
Au
dit
s Fo
urn
isse
urs
La F
DA
: M
issi
on
s, ré
gle
men
tati
on
, pré
par
atio
n e
t d
éro
ule
men
t d
es in
spec
tio
ns
Les
Insp
ecti
on
s Ja
po
nai
ses,
le M
HLW
et
le P
MD
A
RC
/QD
: R
evu
e d
e co
nce
pti
on
/Qu
alifi
cati
on
de
dév
elo
pp
emen
t
Init
iati
on
au
x St
atis
tiq
ues
uti
lisée
s en
val
idat
ion
, co
ntr
ôle
s et
test
s
GxP
Man
ager
: La
ges
tio
n d
es d
on
née
s m
étie
rs, p
roje
ts e
t ré
gle
men
tair
es
Qu
alit
é In
form
atiq
ue
: En
co
nfo
rmit
é av
ec le
s re
com
man
dat
ion
s d
e l’I
TIL
GA
MP
5 : U
tilis
atio
n p
rag
mat
iqu
e d
ans
la m
ise
en c
on
form
ité
rég
lem
enta
ire
Ges
tio
n d
e p
roje
t
Man
agem
ent
/ D
irec
tio
n d
e p
roje
t
An
alys
e d
es b
eso
ins
et c
ahie
r des
ch
arg
es :
Ap
plic
atio
n à
l’in
form
atiq
ue
ou
au
x éq
uip
emen
ts
Man
agem
ent
des
tech
niq
ues
de
test
s in
form
atiq
ue
et re
cett
e d
e lo
gic
iel
L’In
form
atiq
ue
po
ur l
es n
on
sp
écia
liste
s
L’au
tom
atiq
ue
po
ur l
es n
on
sp
écia
liste
s
LIM
S : f
on
ctio
nn
alit
és, c
on
du
ite
du
pro
jet
et v
alid
atio
n
Les
Uti
lités
: Po
ints
clé
s p
ou
r l’e
xpre
ssio
n d
es b
eso
ins
et la
val
idat
ion
De
la G
esti
on
Éle
ctro
niq
ue
de
Do
cum
ents
(GED
) à la
Ges
tio
n E
lect
ron
iqu
e d
e l’I
nfo
rmat
ion
(GEI
)
La M
étro
log
ie :
No
tio
ns
de
bas
e
La m
étro
log
ie d
’un
po
int
de
vue
org
anis
atio
nn
el e
t o
pér
atio
nn
el
A1
A1
0
A1
1
A1
31
A1
5
A4
0
A5
0
B1
0
B2
1
B2
11
B2
20
B2
5
B2
6
B2
7
B2
8
B3
2
B3
3
B3
6
B3
9
B9
B2
9
B3
0
C1
3
C5
8
C5
9
C6
0
C6
1
A1
8
B1
03
B1
04
B8
B3
5
A1
7
A2
0
A3
0
B1
4
B3
4
C1
0
C1
1
C2
0
C2
2
D1
2
D1
3
D1
5
D2
2
D2
6
B1
3
D7
APR (Annual Product Review)
Audit / Inspection
Besoins / URS
Cosmétiques
Dispositifs Médicaux
Enregistrements Electroniques
Equipements
FDA
GAMP
Gestion et direction de projet
Gestion des Risques
ICH
Infrastructure Informatique
Ingénierie
Japon
Laboratoires
Médicaments
Méthodes d’analyse
Métrologie
Nettoyage
Principes Actifs (API)
Règlementation BPx / GxP
Revue de Conception (RC/QD)
Statistiques
Systèmes Automatisés
Systèmes d’Information
Systèmes Qualité
Utilités
Validation
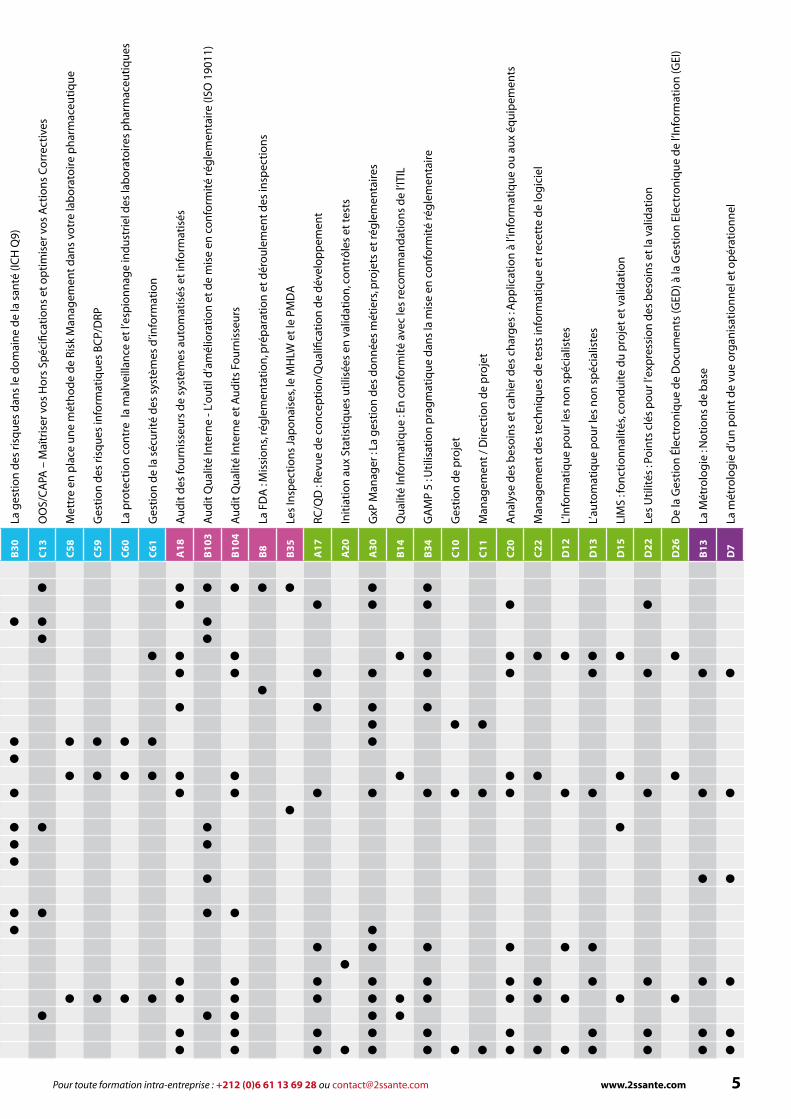
Formations par mots-clés
5Pour toute formation intra-entreprise : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
Sen
sib
ilisa
tio
n à
la v
alid
atio
n
Valid
atio
n d
e sy
stèm
es a
uto
mat
isés
& é
qu
ipem
ents
de
pro
du
ctio
n
Valid
atio
n d
es s
ystè
mes
d’in
form
atio
n
Valid
atio
n d
es m
éth
od
es d
’an
ayse
Valid
atio
n d
es p
rocé
dés
de
net
toya
ge
Qu
alifi
cati
on
d’in
fras
tru
ctu
re In
form
atiq
ue
(Q2I
)
Valid
atio
n d
es s
ystè
mes
et
des
éq
uip
emen
ts d
e la
bo
rato
ire
(VSE
L)
ICH
Q10
: M
ise
en œ
uvr
e p
rag
mat
iqu
e
BPF
et
cGM
P : P
rin
cip
es g
énér
aux,
reto
urs
d’e
xpér
ien
ce e
t n
ou
veau
tés
Form
atio
n a
ctio
n d
es o
pér
ateu
rs s
ur s
ite
aux
BFP
et
GM
P
BPF
Co
smét
iqu
es (I
SO 2
2176
) : P
rin
cip
es e
t o
bje
ctifs
Bo
nn
es p
rati
qu
es d
e la
bo
rato
ire
po
ur l
es e
ssai
s p
récl
iniq
ues
: C
on
form
ité,
AQ
et
Au
dit
Bo
nn
es p
rati
qu
es c
liniq
ues
po
ur l
es e
ssai
s d
e m
édic
amen
t : E
thiq
ue
et q
ual
ité
des
do
nn
ées
Enre
gis
trem
ents
Ele
ctro
niq
ues
et
Ges
tio
n d
es S
ystè
mes
Info
rmat
isés
, il y
a u
n c
on
sen
sus
!
Rég
lem
enta
tio
n d
es d
isp
osi
tifs
méd
icau
x : U
SA F
DA
, Eu
rop
e M
DD
et
mar
qu
age
CE
Rég
lem
enta
tio
n a
pp
licab
le a
ux
pri
nci
pes
act
ifs A
PI /
ICH
Q7
Ass
ura
nce
Qu
alit
é et
Co
ntr
ôle
Qu
alit
é d
ans
les
lab
ora
toir
es d
e co
ntr
ôle
APR
- A
nn
ual
Pro
du
ct R
evie
w /
Rev
ue
Qu
alit
é Pr
od
uit
Les
bo
nn
es p
rati
qu
es d
ans
les
lab
ora
toir
es d
e ch
imie
An
alys
e d
e ri
squ
es C
VO
-RM
(Ris
k M
anag
emen
t)
La g
esti
on
des
ris
qu
es a
pp
liqu
ée a
ux
Dis
po
siti
fs M
édic
aux
(ISO
149
71:2
007)
La g
esti
on
des
ris
qu
es d
ans
le d
om
ain
e d
e la
san
té (I
CH
Q9)
OO
S/C
APA
– M
aîtr
iser
vo
s H
ors
Sp
écifi
cati
on
s et
op
tim
iser
vo
s A
ctio
ns
Co
rrec
tive
s
Met
tre
en p
lace
un
e m
éth
od
e d
e R
isk
Man
agem
ent
dan
s vo
tre
lab
ora
toir
e p
har
mac
euti
qu
e
Ges
tio
n d
es r
isq
ues
info
rmat
iqu
es B
CP/
DRP
La p
rote
ctio
n c
on
tre
la m
alve
illan
ce e
t l’e
spio
nn
age
ind
ust
riel
des
lab
ora
toir
es p
har
mac
euti
qu
es
Ges
tio
n d
e la
séc
uri
té d
es s
ystè
mes
d’in
form
atio
n
Au
dit
des
fou
rnis
seu
rs d
e sy
stèm
es a
uto
mat
isés
et
info
rmat
isés
Au
dit
Qu
alit
é In
tern
e - L
’ou
til d
’am
élio
rati
on
et
de
mis
e en
co
nfo
rmit
é ré
gle
men
tair
e (IS
O 1
9011
)
Au
dit
Qu
alit
é In
tern
e et
Au
dit
s Fo
urn
isse
urs
La F
DA
: M
issi
on
s, ré
gle
men
tati
on
, pré
par
atio
n e
t d
éro
ule
men
t d
es in
spec
tio
ns
Les
Insp
ecti
on
s Ja
po
nai
ses,
le M
HLW
et
le P
MD
A
RC
/QD
: R
evu
e d
e co
nce
pti
on
/Qu
alifi
cati
on
de
dév
elo
pp
emen
t
Init
iati
on
au
x St
atis
tiq
ues
uti
lisée
s en
val
idat
ion
, co
ntr
ôle
s et
test
s
GxP
Man
ager
: La
ges
tio
n d
es d
on
née
s m
étie
rs, p
roje
ts e
t ré
gle
men
tair
es
Qu
alit
é In
form
atiq
ue
: En
co
nfo
rmit
é av
ec le
s re
com
man
dat
ion
s d
e l’I
TIL
GA
MP
5 : U
tilis
atio
n p
rag
mat
iqu
e d
ans
la m
ise
en c
on
form
ité
rég
lem
enta
ire
Ges
tio
n d
e p
roje
t
Man
agem
ent
/ D
irec
tio
n d
e p
roje
t
An
alys
e d
es b
eso
ins
et c
ahie
r des
ch
arg
es :
Ap
plic
atio
n à
l’in
form
atiq
ue
ou
au
x éq
uip
emen
ts
Man
agem
ent
des
tech
niq
ues
de
test
s in
form
atiq
ue
et re
cett
e d
e lo
gic
iel
L’In
form
atiq
ue
po
ur l
es n
on
sp
écia
liste
s
L’au
tom
atiq
ue
po
ur l
es n
on
sp
écia
liste
s
LIM
S : f
on
ctio
nn
alit
és, c
on
du
ite
du
pro
jet
et v
alid
atio
n
Les
Uti
lités
: Po
ints
clé
s p
ou
r l’e
xpre
ssio
n d
es b
eso
ins
et la
val
idat
ion
De
la G
esti
on
Éle
ctro
niq
ue
de
Do
cum
ents
(GED
) à la
Ges
tio
n E
lect
ron
iqu
e d
e l’I
nfo
rmat
ion
(GEI
)
La M
étro
log
ie :
No
tio
ns
de
bas
e
La m
étro
log
ie d
’un
po
int
de
vue
org
anis
atio
nn
el e
t o
pér
atio
nn
el
A1
A1
0
A1
1
A1
31
A1
5
A4
0
A5
0
B1
0
B2
1
B2
11
B2
20
B2
5
B2
6
B2
7
B2
8
B3
2
B3
3
B3
6
B3
9
B9
B2
9
B3
0
C1
3
C5
8
C5
9
C6
0
C6
1
A1
8
B1
03
B1
04
B8
B3
5
A1
7
A2
0
A3
0
B1
4
B3
4
C1
0
C1
1
C2
0
C2
2
D1
2
D1
3
D1
5
D2
2
D2
6
B1
3
D7
APR (Annual Product Review)
Audit / Inspection
Besoins / URS
Cosmétiques
Dispositifs Médicaux
Enregistrements Electroniques
Equipements
FDA
GAMP
Gestion et direction de projet
Gestion des Risques
ICH
Infrastructure Informatique
Ingénierie
Japon
Laboratoires
Médicaments
Méthodes d’analyse
Métrologie
Nettoyage
Principes Actifs (API)
Règlementation BPx / GxP
Revue de Conception (RC/QD)
Statistiques
Systèmes Automatisés
Systèmes d’Information
Systèmes Qualité
Utilités
Validation

VALIDATION / QUALIFICATION

VA
LID
AT
ION
/ Q
UA
LIF
ICA
TIO
N
7Pour toute formation intra-entreprise : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
Le processus de validation doit impérativement être porté par les structures dirigeantes de l’industrie. La formation Sensibilisation à la validation, particulièrement adaptée à cette population, permet de résumer les enjeux de la validation. Au-delà des contraintes réglementaires, les apports de la validation sont aujourd’hui incontestables. Amélioration de la maîtrise des systèmes, diminution des risques projets et des coûts de maintenance, sont autant de raisons qui font de la validation, un outil indispensable à tout projet de système.
Sensibilisation à la validation*
Programme
● Comprendre les différentes stratégies de
validation pour les systèmes informatisés,
équipements et procédés d’une entreprise
ou d’un site industriel.
● Savoir définir une stratégie de validation à
l’échelle d’un site, d’un pays, d’un groupe.
● Savoir adapter sa stratégie de validation en
fonction de la maturité de l’organisation.
● Faire le point sur les évolutions des
exigences réglementaires de l’AFSSAPS,
ou EMEA et/ou de la FDA applicables à
la validation.
● Savoir déterminer l’effort et le retour sur
investissement d’une validation bien
conduite.
● Connaître le contenu d’un dossier de
validation.
Objectifs
* Cette formation peut être réalisée sur une durée de deux heures à une journée ; elle peut aussi être adaptée pour vos besoins spécifiques.
A1
PUBLIC
Etant modulable et adaptable, cette
formation peut s’adresser au manage-
ment de l’entreprise sous forme de sen-
sibilisation ou tout autre public devant
acquérir les bases, les définitions ainsi que
les justifications de la validation.
A11 A40
A131 A50
A15
Pour développer vos compétences, nous vous conseillons également :
A10
1. IntroductionDéfinition de la validation. Le but de la
validation. Les référentiels réglementaires.
Comprendre la nécessité de valider à partir
des textes applicables à vos installations : BPF,
cGMP, BPL, BPC, 21 CFR Part 11, ICH.
2. Stratégie de validationMise en place d’une politique de validation.
Détermination du niveau de maturité de
votre entreprise à l’aide du VMM (Validation
Maturity Model) de CyberConseil. Les étapes,
les concepts et les démarches de validation.
Type de validation applicable à vos systèmes.
Activités de validation à planifier, à réaliser et
à optimiser.
3. Les erreurs à éviter : étude de cas réelsPrésentation et discussion sur les «Warning
Letter» et «483» afin de comprendre ce que
les agences réglementaires attendent au
niveau de la validation des procédés, des
équipements, des méthodes analytiques et
des systèmes informatisés et automatisés.
4. Les étapes de qualificationPrésentation des points-clés d’un dossier de
validation. Familiarisation avec la Qualification
Fournisseur, la Qualification de Conception,
la Qualification d’Installation, la Qualification
Opérationnelle et la Qualification de Perfor-
mance. Gestion et suivi de l’état de validation
de vos systèmes.
5. Retour d’expériences et statistiquesRetour d’expériences au travers d’exemples
de projets réussis. Statistiques sur les coûts et
les gains de la validation.
6. Préparation d’une inspectionPoints-clés de la préparation d’une inspection
de l’AFSSAPS ou de la FDA.
Evaluation des connaissances acquises et
conclusion du séminaire.
Un QCM vous est proposé afin d’évaluer les
connaissances acquises.
Vérification de l’atteinte des objectifs établis
en début de séminaire.
B8
Toutes nos formations sont
réalisables en intra-entreprise
Contactez-nous + 212 (0)6 61 13 69 28
www.2ssante.com

8
VA
LID
AT
ION
/ Q
UA
LIF
ICA
TIO
N
Pour tout projet de formation : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
Les systèmes automatisés et les équipements de production sont, pour la plupart, en contact physique avec les produits, et ainsi, traditionnellement exposés aux inspections. Les impacts d’une validation inadaptée sont donc extrêmement coûteux, tant en termes d’image que d’impact produit. Les systèmes devenant de plus en plus complexes, la récupération des tests statiques et dynamiques des concepteurs et fournisseurs pour supporter les QI et QO devient cruciale mais encore faut-il que ces tests soient récupérables et acceptables d’un point de vue réglementaire.
1. Points clés et enjeux
1.1. DéfinitionsDéfinitions et objectifs de la validation.
1.2. Les organismesPrésentation et description des principales instances réglementaires et normatives.
1.3. Les réglementations, les guides et les normesDescription des exigences et propositions des référentiels français, européens, nord-américains et internationaux : BPF, cGMP, ICH, 21 CFR, GAMP, PIC/S, normes ISO-NF.Position des agences officielles. Présentation de Warning Letters et/ou 483s de la FDA.
1.4 Les principes de baseComment justifier la nécessité de valider vos installations et vos équipements ?Concepts d’indépendance et de points critiques.
2. Validation et MaturitéPrésentation du VMM (Validation Maturity Model).Comment déterminer le niveau de maturité de votre entreprise ?Impacts de la maturité sur les projets de validation.
3. Détermination de la Stratégie de Valida-tion et des systèmes à valider – Plan Direc-teur de Validation (PDV)
3.1. Stratégie de ValidationMaîtriser les étapes, les concepts et les dé-marches de la validation.Validation prospective, rétrospective, conco-mitante.Les principales activités de validation et de qualification.Différencier la validation des équipements et la validation des procédés.
3.2. Plan Directeur de Validation (PDV)Présentation de l’objectif et de l’organisation d’un VMP. Définir le type de validation / qualification applicable à vos équipements et systèmes automatisés.Comment obtenir un VMP précis, non am-bigu et cohérent, ainsi que la matrice des systèmes ?Comment gérer et suivre l’état de validation de vos systèmes ?
4. Détermination des activités de valida-tion pour un système ou un groupe de systèmes - Plan de Validation (PV)Définition et objectifs du plan de validation.Que doit contenir un plan de validation ?
PÉRIMÈTRE
• Equipements de production : cuves, réacteurs, étuves, …
• Systèmes automatisés de production : remplisseuse, blistériseuse, centrale de pesée, …
• Logiciels de production : étiquetage, enregistrement de données,…
• Utilités : GTC, GCM, Eaux,…
• Equipements de mesure : balances, dynamomètre,…
• Logiciels de Maintenance et de Métro-logie : GMAO, …
Validation des Systèmes Automatiséset des Equipements de Production (VSAEP)
Programme
● Savoir définir une stratégie de validation adaptée à une situation particulière (mono produit, multi produit, installation auto-matisée ou non, validation prospective ou non) et au contexte projet (maturité des fournisseurs, bonnes pratiques…).
● Connaître les exigences réglementaires et normatives applicables à la validation et déterminer les informations du dossier de validation indispensables pour être en conformité avec les exigences régle-mentaires et les guides de l’AFSSAPS, de l’EMEA et/ou de la FDA (BPF/GMP, BPL/GLP, BPC/GCP, QSR, ICH, PIC/S, ISO…).
● Savoir définir et optimiser l’effort de vali-dation nécessaire en fonction du contexte et répartir les activités de validation entre les différents acteurs.
● Avoir les éléments de base pour définir et mettre en œuvre un Plan Directeur
de Validation (PDV/VMP), les Plans de Validation (PV), les Qualifications Four-nisseur (QF), les Revues de Conception / Qualifications de Développement (RC/QD), les analyses des risques (CVO-RM©), les Qualifications d’Installation, Opéra-tionnelle et de Performance (QI, QO, QP), les matrices de traçabilité.
● Connaître l’étendue des techniques nécessaires pour réussir la validation d’un système industriel.
● Distinguer les tests de recette/FAT/SAT des tests de validation et savoir intégrer les tests fournisseur dans la validation (Validation Intégrée).
● Savoir utiliser les recommandations du GAMP 5 de façon pragmatique.
Objectifs Formation inter-entreprises
DUREE 3 jours
A10FORMATION ACTUALISEE
PUBLIC
- Responsable Qualité,
- Responsable Validation/Qualification
- Equipes d’ingénierie
- Equipes de maintenance/métrologie
- Equipes de qualification
A11 A40 A15
A131 A50 A1
Pour développer vos compétences, nous vous conseillons également :
B34D13

VA
LID
AT
ION
/ Q
UA
LIF
ICA
TIO
N
9Pour toute formation intra-entreprise : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
Comment définir l’organisation, les respon-sabilités, choisir la démarche et les activités de validation dans un Plan de Validation co-hérent et non ambigu pour chacun de vos systèmes ou groupes de systèmes.Comment déterminer les activités de qualifi-cation à mettre en œuvre ?Doit-on toujours faire des QF, QD, QI, QO, QP ? Sur quoi se baser pour justifier ses choix ?Cas pratique étape 1/6.
5. Qualification des Fournisseurs (QF) Définition et objectifs de la QF.Les points principaux à prendre en compte ; les questions à ne pas oublier ; les textes de références utilisables.Comment utiliser l’audit fournisseur dans un esprit « Validation Intégrée » afin de faciliter la récupération des tests fournisseurs pour soutenir la QI et la QO.
6. Revue de Conception (RC) ou Qualifica-tion de Développement (QD)Définition et objectifs de la QD.Présentation des principales méthodes pour la formaliser. Détail de la Revue de Conception permettant de prouver la prise en compte de la régle-mentation et des bonnes pratiques d’ingé-nierie lors de la conception de vos systèmes.Quand et Qui fait la revue de code ?Initialisation de la matrice de traçabilité (des exigences aux spécifications de conception).Cas pratique étape 2/6.
7. Détermination des éléments à qualifier et des cas de tests à mettre en œuvre - Analyse de risques (AR) Définition et présentation des objectifs de l’Analyse de Risques.Rappel des concepts de base et de la théorie de l’analyse de risques.Comment définir les risques ? Comment définir une bonne échelle de risques ?Comment définir des tests pertinents à partir des résultats de l’analyse de risques ?Relations entre Qualification de Développe-ment (QD) et Analyse de Risques.Mise à jour de la matrice de traçabilité (liens Risques – Composants / Fonctions / Proces-sus – liste des tests).Présentation de la méthode d’analyse de risques CVO-RM.Cas pratique étape 3/6.
8. Qualification d’Installation (QI)Définition et objectifs de la QI. Ce qu’est et ce que n’est pas la QI. Pré-requis indispensables à toute QI. Que doit-il y avoir dans une QI ?Quand doit-on vérifier la métrologie ?
Les fiches de test de QI.Rédaction des cas de test de QI déterminés par le résultat de l’analyse de risques composant.Comment capitaliser sur la QI et construire des protocoles standards ?Mise à jour de la matrice de traçabilité (liens Risques – Composants – Cas de test).Cas pratique étape 4/6.
9. Qualification Opérationnelle (QO)Définition et objectifs de la QO.Pré-requis indispensables à toute QO.Définition des différents types de test de qualification opérationnelle : Nominal, Limite (limite des champs, des paramètres et des spé-cifications matières ; cas le plus défavorable ; charge ; stress), Défaillant (situation anormale de fonctionnement).Les fiches de test de QO.Rédaction des cas de test de QO déterminés par le résultat de l’analyse de risques fonctionnelle.Tests fournisseurs versus tests de QO. Quand peut-on récupérer les tests fournisseurs (FAT / SAT ; recette) ?Comment déterminer les Cas les plus défavo-rables (Worst Cases) ?Mise à jour de la matrice de traçabilité (liens Risques – Fonctions – Cas de test).Cas pratique étape 5/6.
10. Qualification de Performance (QP)Définition et objectifs de la QP.Domaine d’application de la QP.Pré-requis indispensables à toute QP.Les trois phases de la QP : QPA - Vérification documentaire avant mise production ; QPS - Simulation avant utilisation de produit ; QPR - Routine et Reproductibilité.Les fiches de test de QP.Rédaction des cas de test de QP déterminés par le résultat de l’analyse de risques processus.Est-ce que 3 lots sont toujours nécessaires et/ou suffisants ?Les spécificités des QP logiciels.Mise à jour de la matrice de traçabilité (liens Risques – Processus – Cas de test).Cas pratique étape 6/6.
11. Conduite des tests, rapports et exploi-tationFamiliarisation avec le déroulement des tests.La documentation et la gestion des preuves. Mise à jour de la matrice de traçabilité (liens Risques – Composants / Fonctions / Processus – Cas de test – Résultats de l’exécution - Ecarts).Le contenu des rapports de validation.Maintenir un système validé : La clé du succès réside dans la maîtrise des change-ments : Change Control versus Change Ma-nagement. Evaluations périodiques versus Revalidation.
Comment gérer la non-régression suite aux modifications.Mise à jour de la matrice de traçabilité (liens Risques – Composants / Fonctions / Proces-sus – Cas de test – Modifications).
12. Gestion de la documentation Comment gérer tous ces documents de vali-dation à établir ?Comment mettre en place un référentiel validation, une structure documentaire et une organisation performante ?Quelles sont les règles documentaires de base qui permettent d’assurer une traçabilité suffisante de vos réalisations ?Relations entre les différents documents générés par les utilisateurs, l’ingénierie, la validation.Rappel sur les bonnes pratiques documen-taires.
EXERCICES DE MISE EN SITUATIONUn cas pratique en forme de fil rouge sur un ou plusieurs thèmes selon le nombre de participants et leurs préférences est déroulé tout au long de cette formation ; les étapes concernées sont :- 4. Plan de Validation- 6. Qualification de Développement- 7. Analyse de Risques- 8. Qualification d’Installation- 9. Qualification Opérationnelle- 10. Qualification de Performance
Un QCM vous est proposé afin d’évaluer les connaissances acquises.
Validation des Systèmes Automatiséset des Equipements de Production (VSAEP)
A10FORMATION ACTUALISEE
A18
A17
B9
D26
Toutes nos formations sont
disponibles en anglais
Contactez-nous + 212 (0)6 61 13 69 28
www.2ssante.com

10
VA
LID
AT
ION
/ Q
UA
LIF
ICA
TIO
N
Pour tout projet de formation : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
y Connaître les exigences réglementaires et normatives applicables à la validation. Comprendre l’environnement réglemen-taire pour être en conformité avec les exigences de l’AFSSAPS, de l’EMEA et/ou de la FDA (BPF/GMP, BPL/GLP, BPC/GCP, QSR…).
y Organiser les validations à l’échelle d’un site, d’un pays, d’un groupe et mettre en œuvre un Plan Directeur de Validation (PDV/VMP).
y Savoir définir une stratégie de validation adaptée à un environnement informa-tisé (validation prospective ou non) et au contexte projet (maturité des fournisseurs, bonnes pratiques…).
y Connaître l’étendue des enjeux et com-prendre les techniques nécessaires pour réussir la validation d’un système d’infor-mation : le Plan de Validation (PV), l’analyse de risques (CVO-RM© et les techniques classiques), les qualifications d’installation, opérationnelle et de performance (QI, QO, QP), les rapports de validation, la gestion
des modifications. Appliquer le « cycle en V ».
y Comprendre l’importance de l’ingénie-rie des besoins dans le processus de validation : Le Cahier des Charges d’un système d’information est essentiel pour valider : points-clés, démarche et contenu. Qu’attendre des Spécifications Fonction-nelles ? Comment construire un dossier de conception complet. Savoir intégrer les tests fournisseur dans la validation (Vali-dation Intégrée).
y Savoir être critique vis à vis de ces docu-ments d’ingénierie : conduire les Revues de Conception (RC aussi appelées QD).
y Savoir définir et optimiser l’effort de vali-dation nécessaire en fonction du contexte et répartir les activités de validation entre les différents acteurs (services validation, qualité, informatique, ingénierie, …).
y Savoir utiliser les recommandations du GAMP 5 de façon pragmatique.
De plus en plus complexes, les systèmes d’information prennent une place croissante dans les entreprises. Bien que n’étant pas en contact direct avec les produits, ils permettent de gérer des informations hautement critiques (caractéristiques, traçabilité, statuts, …). Pour plus de sécurité, pour diminuer les coûts et afin d’être en conformité avec les réglementations internationales, la validation des systèmes d’information est indispensable dans les industries pharmaceutiques, chimiques, cosmétiques et pour les dispositifs médicaux.
PUBLIC
- Responsable Assurance Qualité,
- Responsable Validation / Qualification
- Responsable Informatique
- Chef de Projet utilisateur ou
informatique
- Key User / Utilisateur référent
1. Points clés et enjeux
1.1. DéfinitionsDéfinitions et objectifs de la validation.
1.2. Les organismesPrésentation et description des principales instances réglementaires et normatives.
1.3. Les réglementations, les guides et les normesDescription des exigences et propositions des référentiels français, européens, nord-américains et internationaux : GMP, GLP, GCP, 21 CFR Part 11, BPF, BPL, BPC, ICH, GAMP, PIC/S, normes ISO-NF.Position des agences officielles. Points-clés d’une inspection. Analyse des textes à l’aide de Warning Letters et/ou de 483’s de la FDA.
1.4 Les principes de baseIntroduction à la validation des systèmes d’information.Concepts d’indépendance et de points cri-tiques.
2. Validation et MaturitéPrésentation du VMM (Validation Maturity Model).Comment déterminer le niveau de maturité de votre entreprise ?Impacts de la maturité sur les projets de validation.
3. Détermination de la Stratégie de Valida-tion et des systèmes à valider – Plan Direc-teur de Validation (PDV)
3.1. Stratégie de ValidationComment définir et déployer une politique de validation. Présentation, avec des exemples concrets, de la démarche de validation. Détermination du «juste nécessaire» pour valider un système d’information. Identification des facteurs déterminants pour la réussite d’un projet de validation.Maîtriser les étapes, les concepts et les dé-marches de la validation.Les principales activités de validation et de qualification.
PÉRIMÈTRE
• ERP
• Gestion et analyse de données cliniques
• Pharmacovigilance
• GED
• LIMS
• Macro Excel
• Développement Access
Programme
Objectifs
Validation des Systèmes d’Information FORMATION ACTUALISEE
A11
Formation inter-entreprises
DUREE 3 jours
A10 A40 A50
A15 A131 A1
Pour développer vos compétences, nous vous conseillons également :
D12 B34

VA
LID
AT
ION
/ Q
UA
LIF
ICA
TIO
N
11Pour toute formation intra-entreprise : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
3.2. Plan Directeur de Validation (PDV)Présentation de l’objectif et de l’organisation d’un VMP. Définir un «Validation Master Plan» pour le pilotage de vos validations.Comment obtenir un VMP précis, non ambigu et cohérent, ainsi que la matrice des systèmes ? Comment gérer et suivre l’état de validation de vos systèmes ?Cas pratique étape 1/6.
4. Détermination des activités de validation pour un système ou un groupe de systèmes - Plan de Validation (PV)Définition et objectifs du plan de validation.Que doit contenir un plan de validation ?Comment définir l’organisation, les respon-sabilités, choisir la démarche et les activités de validation dans un Plan de Validation co-hérent et non ambigu pour chacun de vos systèmes ou groupes de systèmes.Comment déterminer les activités de qualifi-cation à mettre en œuvre ?Doit-on toujours faire des QF, QD, QI, QO, QP ? Sur quoi se baser pour justifier ses choix ?Cas pratique étape 2/6.
5. Qualification des Fournisseurs (QF) Définition et objectifs de la QF.Les points principaux à prendre en compte ; les questions à ne pas oublier ; les textes de ré-férences utilisables. Faut-il auditer avant ou après le choix de l’outil ? Comment utiliser l’audit fournisseur dans un esprit « Validation Intégrée » afin de faciliter la récupération des tests fournisseurs pour soutenir la QI et la QO.
6. Revue de Conception (RC) ou Qualifica-tion de Développement (QD) Définition et objectifs de la QD.Présentation des principales méthodes pour la formaliser. Détail de la Revue de Conception permettant de prouver la prise en compte de la réglemen-tation et des bonnes pratiques d’ingénierie lors de la conception de vos systèmes. Quand et Qui fait la revue de code ? Initialisation de la matrice de traçabilité (des exigences aux spécifications de conception).
7. Détermination des éléments à qualifier et des cas de tests à mettre en œuvre - Analyse de risques (AR) Définition et présentation des objectifs de l’Analyse de Risques.Rappel des concepts de base et de la théorie de l’analyse de risques.Comment définir les risques ? Comment définir une bonne échelle de risques ?Comment définir des tests pertinents à partir des résultats de l’analyse de risques ?Relations entre Qualification de Développe-
ment (QD) et Analyse de Risques.Mise à jour de la matrice de traçabilité (liens Risques – Composants / Fonctions / Proces-sus – liste des tests).Présentation de la méthode d’analyse de risques CVO-RM.Cas pratique étape 3/6.
8. Qualification d’Installation (QI)Définition et objectifs de la QI. Ce qu’est et ce que n’est pas la QI. Pré-requis indispensables à toute QI. Que doit-il y avoir dans une QI ?Les fiches de test de QI.Rédaction des cas de test de QI déterminés par le résultat de l’analyse de risques composant.Comment traiter les ressources communes (Salle machine, réseau, sauvegarde et restau-ration, …) et passer de la validation verticale à la validation horizontale ?Comment capitaliser sur la QI et construire des protocoles standards ?Mise à jour de la matrice de traçabilité (liens Risques – Composants – Cas de test)Cas pratique étape 4/6.
9. Qualification Opérationnelle (QO)Définition et objectifs de la QO.Pré-requis indispensables à toute QO. Qu’est ce qu’une bonne spécification fonctionnelle ?Définition des différents types de test de qualification opérationnelle : Nominal, Limite (limite des champs ; cas le plus défavorable ; stress), Défaillant (situation anormale de fonc-tionnement).Les fiches de test de QO.Rédaction des cas de test de QO déterminés par le résultat de l’analyse de risques fonc-tionnelle.Dans quel cadre et avec quelles précautions utiliser les tests des fournisseurs ? Savoir les vérifier. Leurs avantages et leurs limites.Mise à jour de la matrice de traçabilité (liens Risques – Fonctions – Cas de test)Cas pratique étape 5/6.
10. Qualification de Performance (QP)Définition et objectifs de la QP.Domaine d’application de la QP.Pré-requis indispensables à toute QP. En quoi est-ce important d’avoir un bon Cahier des Charges ?Les trois phases de la QP : QPA - Vérification documentaire Avant mise production ; QPS - Simulation avant utilisation sur des données réelles ; QPI - Indicateurs + Reproductibilité.Les fiches de test de QP.Rédaction des cas de test de QP déterminés par le résultat de l’analyse de risques processus.Les spécificités des QP logiciels.Mise à jour de la matrice de traçabilité (liens
Risques – Processus – Cas de test).Cas pratique étape 6/6.
11. Conduite des tests, rapports et exploi-tationFamiliarisation avec le déroulement des tests.La documentation et la gestion des preuves. Mise à jour de la matrice de traçabilité (liens Risques – Composants / Fonctions / Processus – Cas de test – Résultats de l’exécution - Ecarts)Le contenu des rapports de validation.Maintenir un système validé : La clé du suc-cès réside dans la maîtrise des changements (Change Control). Pratiques indispensables pour assurer la validité de vos systèmes dans le temps. Quand revalider et jusqu’à quel niveau de détail ?Comment gérer la non-régression suite aux modifications.Mise à jour de la matrice de traçabilité (liens Risques – Composants / Fonctions / Processus – Cas de test – Modifications)
12. Gestion de la documentation Comment gérer tous ces documents de vali-dation à établir ?Comment mettre en place un référentiel validation, une structure documentaire et une organisation performante ?Quelles sont les règles documentaires de base qui permettent d’assurer une traçabilité suffisante de vos réalisations ?Relations entre les différents documents générés par les utilisateurs, l’ingénierie, la validation.Rappel sur les bonnes pratiques documen-taires.
EXERCICES DE MISE EN SITUATION
Un cas pratique en forme de fil rouge sur un système type ERP est déroulé tout au long de cette formation ; les étapes concernées sont :- 3. Plan Directeur de Validation- 4. Plan de Validation- 7. Analyse de Risques- 8. Qualification d’Installation- 9. Qualification Opérationnelle- 10. Qualification de Performance
Un QCM vous est proposé afin d’évaluer les connaissances acquises.
Validation des Systèmes d’InformationA11
FORMATION ACTUALISEE
A18
A17
B9
D26

12
VA
LID
AT
ION
/ Q
UA
LIF
ICA
TIO
N
Pour tout projet de formation : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
Dans un environnement réglementaire (BPF, BPL, cGMP et ICH), la validation des méthodes est, avec la qualification des équipe-ments, à la base de la qualité des données analytiques. Processus décrit et strictement encadré par des textes réglementaires, la mise en place d’une démarche de type : Plan de Validation, rédaction de protocoles, de fiches de tests et du Rapport de Valida-tion, est incontournable. Les analyses réalisées dans le cadre du dossier d’AMM, des contrôles de routine, des études de stabilité, des analyses d’impuretés, doivent être validées selon des critères pré-définis : spécificité, détectabilité, sensibilité, exactitude, reproductibilité, répétabilité, et à l’aide d’outils statistiques.
1. Positionnement et Contexte réglemen-taireImportance des méthodes d’analyses à travers
l’étude des flux analytiques de la Recherche &
Développement et du Contrôle Qualité.
Historique de la validation analytique.
Référentiels réglementaires et normatifs :
Description des exigences et propositions
des référentiels français, européens, nord-
américains et internationaux : BPF, cGMP, BPL,
GLP, Pharmacopées (PE, USP, JP) et ICH.
Définition des critères de validation en fonc-
tion du type de méthode d’analyses et de
leur domaine d’application.
2. Démarche de validation des méthodes d’analysesOrganisation : Analyse de l’existant afin de
définir les priorités de validation des mé-
thodes d’analyses, rédaction des plans de
Validation, positionnement des validations
de techniques Analytiques.
Organisation : Définition et analyse des élé-
ments clés participant à la validation.
Stratégies : Démarche de recherche d’équiva-
lence entre différentes méthodes et transfert
de méthodes d’analyses au sein de plusieurs
laboratoires.
Documentation : Techniques de rédaction de
protocoles de validation, de fiches de tests, de
rapports de validation.
3. Outils utilisés lors de la validation de méthodes d’analysesLes différents types de calculs statistiques
disponibles et utilisés.
Les règles à respecter sur l’utilisation des pro-
duits existants sur le marché ou développés
en interne.
4. Etude de casDéroulement d’une validation de la méthode
d’analyse et interprétation.
5. Suivi de validationRevalidation de méthodes d’analyses, vérifi-
cation d’aptitude, retour d’expérience.
EXERCICES DE MISE EN SITUATIONPlusieurs exercices sont proposés pendant la
formation pour illustrer les points abordés.
Un QCM vous est proposé afin d’évaluer les
connaissances acquises.
Validation des méthodes d’analyse
Programme
● Connaître la réglementation applicable :
française, européenne, américaine et ICH
en matière de validation analytique.
● Savoir définir et mettre en place une
démarche de validation des méthodes
d’analyses en fonction des situations
(Nouvelle méthode analytique, équiva-
lence de méthode, transfert de méthode).
● Etre capable d’exploiter et d’interpréter
les résultats d’une validation de méthodes
d’analyses.
Objectifs
A131
PUBLIC
- Correspondant Assurance Qualité pour
le laboratoire
- Responsable de laboratoire
- Technicien de laboratoire chargé de
validation
- Equipes de Qualification / Validation
pour le laboratoire
A11 A40 A15
A50 A1
Pour développer vos compétences, nous vous conseillons également :
A10
Vous souhaitez obtenir
plus d’informations ?
Contactez-nous + 212 (0)6 61 13 69 28
www.2ssante.com

VA
LID
AT
ION
/ Q
UA
LIF
ICA
TIO
N
13Pour toute formation intra-entreprise : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
Pour être en conformité avec les réglementations internationales applicables aux industries pharmaceutiques, chimiques, cosmétiques et des dispositifs médicaux, la validation des procédés de nettoyage est indispensable. Afin de valider correctement ces procédés, il faut tout d’abord bien les comprendre et ensuite s’assurer que certains pré-requis indispensables sont satisfaits.
Module 1 : Le Contexte réglementaireConnaître les exigences réglementaires concernant le nettoyage et sa validation : BPF (AFSSAPS), cGMP (FDA), GMP (EMA). Connaître les normes concernant le nettoyage et sa validation. Définition des termes employés dans la mise en œuvre et l’utilisation de pro-cédés de nettoyage et la validation de ces procédés.
Module 2 : Les pré-requis à la validation d’un procédé de nettoyage
1. Choix et qualification des méthodes de nettoyage et des détergentsRappel des principales méthodes de net-toyage. Rappel des principales techniques de nettoyage. Rappel des principaux systèmes de nettoyage. Les outils de choix des mé-thodes de nettoyage et des agents de net-toyage associés.Choix des agents de nettoyage :- eau de nettoyage- solvant- détergentChoix des agents de désinfection. Définir les actions préalables à la validation du procédé de nettoyage. Comment choisir et qualifier le matériel de nettoyage. Cas pratique : rédaction d’un cahier des charges pour le choix d’un dé-tergent.
2. Rédaction des procédures de nettoyageLes différentes stratégies de nettoyage po-ssibles selon le nombre et les types de pro-duits. Les risques liés au choix de la stratégie de nettoyage. Les points fondamentaux à inclure dans une procédure de nettoyage complète et applicable aux produits et aux installations. Les risques liés au non suivi de la procédure. La structure d’une procédure
de nettoyage. Cas pratique : rédaction d’une procédure de nettoyage.
3. Méthodes d’analyses à développer et à valider Rappel des principales méthodes d’analyse utilisables. Sélection des méthodes analy-tiques adaptées au contexte. Développer les différentes méthodes d’analyses dans le cadre des procédés de nettoyage pour le dosage des traces de principes actifs, des solvants et des détergents. Le pourcentage de récupération. Méthode de dosage des traces.
Module 3 : La validation du procédé de nettoyage
1. Stratégie de validation des nettoyages : Matrice Equipements/ProduitsL’approche matricielle. Savoir regrouper les produits / équipements afin de limiter le nombre de tests (bracketing). Savoir détermi-ner le(s) pire(s) des cas.
2. Plan d’échantillonnageComment déterminer le nombre de prélève-ments. Comment déterminer les points cri-tiques de prélèvements :- Difficulté de nettoyage- Importance des salissures- Examen des P&ID
3. Méthodes et matériels de prélèvement des échantillonsRappel des différentes méthodes de prélève-ment :- Essuyage ou écouvillonnage (SWAB)- Contact - …Les supports de prélèvement. Les solvants de prélèvement. Les solvants de rinçage.
Avantages et inconvénients de la méthode par SWAB. Avantages et inconvénients de la méthode du solvant. Savoir choisir entre un prélèvement de surface ou un prélèvement de solutions de rinçage. L’importance du protocole. Choisir et qualifier le matériel de prélèvement.
4. Critères d’acceptationConnaître les différents critères : visuels, chimiques et microbiologiques. Le niveau de propreté. Calculer les limites résiduelles acceptables selon le type de produits concernés (produits injectables, produits topiques...). Déterminer et justifier des cri-tères d’acceptation précis, non ambigus et cohérents :- A partir d’une dose journalière – MACO- A partir de données toxicologiques – NOEL- Par aire de surface – LRALa méthode du TOC – Total Organic Carbon. Avantages et inconvénients des méthodes de calcul
5. Documentation de validationContenu et points-clés du protocole de vali-dation. Contenu du rapport de validation de nettoyage. Quelles sont les attentes des ins-pecteurs ?
EXERCICES DE MISE EN SITUATION A PAR-TIR D’EXEMPLES CONCRETS Détermination des « worst case » équipe-ments, procédures de nettoyage, produits.Choix des critères d’acceptation.Un QCM vous est proposé afin d’évaluer les
connaissances acquises.
Validation des procédés de nettoyage
Programme
● Connaître les exigences des réglemen-tations BPF de l’AFSSAPS, cGMP de la FDA et GMP de l’Europe et des normes internationales concernant les pratiques et principes de nettoyage ainsi que les méthodes de validation.
● Savoir mettre en place la validation des procédés de nettoyage et les procédures de nettoyage associées.
● Savoir mettre en place les pré-requis à la validation de nettoyage.
● Savoir définir l’effort de validation en fonction des caractéristiques des produits ainsi que de l’organisation de la fabrication et du nettoyage.
● Savoir déterminer la ou les méthodes de prélèvement et le plan d’échantillonnage associé.
● Savoir définir les limites résiduelles accep-tables.
Objectifs
A15
Formation inter-entreprises
FORMATION ACTUALISEE
PUBLIC
- Assurance Qualité
- Contrôle Qualité
- Production
- Qualification / Validation
DUREE 2 jours
A131
A20

14
VA
LID
AT
ION
/ Q
UA
LIF
ICA
TIO
N
Pour tout projet de formation : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
Les infrastructures informatiques supportent toutes les applications critiques de votre entreprise et elles doivent donc être qualifiées pour plus de sécurité et afin d’être en conformité avec les réglementations internationales.Cette formation fait une place importante à la pratique en ne présentant que l’essentiel d’un point de vue théorique et en intégrant des études de cas et des exercices de mise en situation permettant de comprendre comment passer de la théorie à la pratique sur plusieurs types d’éléments d’infrastructure informatique.
sites, multi-pays.
7. La qualification systématiqueLe minimum requis d’un point de vue orga-nisationnel pour supporter efficacement une qualification d’infrastructure informatique.Quelle organisation mettre en œuvre pour supporter les qualifications et pourquoi ?Présentation d’une approche permettant d’inclure la qualification dans les processus opérationnel de l’infrastructure.Quand et pourquoi remplacer les protocoles de qualification par des procédures de qua-lification ?
8. La gestion des changements sur l’infras-tructurePrésentation des approches possible pour gérer les changements sur l’infrastructure.Comment intégrer les aspects réglementaires sur un processus de gestion des change-ments « ITIL » ?
9. Conduite des tests et rapports et exploi-tationFamiliarisation avec le déroulement des tests et le contenu des rapports de validation et de qualification.
EXERCICES DE MISE EN SITUATION
- Etude d’une approche de qualification pour une infrastructure complexe
- Plan de qualification
Un QCM vous est proposé afin d’évaluer les connaissances acquises.
1. IntroductionRappels sur la Validation : Définitions et ob-jectifs de la validation. Position des agences officielles. Référentiels réglementaires et normatifs: Description des exigences et propositions des référentiels français, européens, nord-américains et internationaux : BPF, cGMP, BPL, GLP, ICHQ7A, 21 CFR Part 11, GAMP, normes ISO-NF, ITIL, …. Principes de validation et de qualification : Les documents de validation à établir ainsi que les règles de base et les bonnes pratiques documentaires qui per-mettent d’assurer une traçabilité suffisante de vos réalisations.
2. Les étapes de qualificationPrésentation / Rappel sur les étapes de qua-lification : - QF : Familiarisation avec la Qualification Fournisseur - QF (prouver que les éléments sont conçus et réalisés selon les bonnes pratiques techniques et réglementaires en vigueur).
- QD : Familiarisation avec la Qualification de Développement - QD (prouver que toutes les exigences utilisateurs et réglementaires ont été correctement prises en compte).
- QI : Familiarisation avec la Qualification d’Installation - QI (prouver la bonne instal-lation de vos systèmes).
- QO : Familiarisation avec la Qualification Opérationnelle - QO (prouver le bon fonctionnement de vos systèmes à l’aide de tests nominaux, défaillants et limites / Worst Case).
- QP : Familiarisation avec la Qualification de
Performance - QP (prouver la fiabilité et le service régulier des systèmes).
3. Qualification Master PlanPrésentation de l’objectif et de l’organisation d’un Qualification Master Plan. Comment déterminer les stratégies de qualification ? Comment obtenir un Qualification Master Plan précis, non ambigu et cohérent ? Comment découper l’infrastructure informa-tique en « briques » qualifiables séparément ?
4. Plan de QualificationPrésentation de l’objectif et des méthodo-logies de qualification possibles. Comment définir l’organisation, les responsabilités, choisir la démarche et les activités de qualifi-cation dans un Plan cohérent et non ambigu pour chaque « brique » d’infrastructure ?
5. Etudes de cas : La Salle Machine : Présentation des straté-gies de qualification possibles.Exemple de qualification de salle machine (incluant HVAC, Onduleur….) Le Réseau : Présentation des stratégies de qualification possibles, problématique des systèmes ouverts/fermé. Exemple de qualification d’un réseau com-plexe.
6. Exemple de qualification : Les systèmes partagés (SAN ; Plateforme de virtualisa-tion…)Présentation des stratégies de qualification possibles, problématique des infrastructures logiques, virtualisées ; problématiques multi-
Qualification d’Infrastructure Informatique (Q2I)
Programme
● Connaître les exigences réglementaires et normatives applicables à l’infrastructure informatique et déterminer les informa-tions indispensables pour être en confor-mité avec les exigences réglementaires de l’AFSSAPS, de l’EMEA et/ou de la FDA (BPF/GMP, BPL/GLP, BPC/GCP, QSR…).
● Savoir identifier les éléments critiques de l’infrastructure nécessitant des actions de qualifications.
● Savoir définir une stratégie et des mé-thodologies de qualification adaptée à chaque « brique » et plateforme d’in-frastructure et au contexte projet (matu-rité des fournisseurs, bonnes pratiques…)
afin d’optimiser l’effort nécessaire pour assurer la conformité réglementaire de l’infrastructure.
● Connaître les éléments clés de l’organisa-tion et du système qualité informatique qui permettent de réaliser et de suppor-ter efficacement la qualification d’infras-tructure informatique.
● Savoir qualifier une infrastructure infor-matique en conformité avec les recom-mandations GAMP IT.
● Savoir définir les processus clef à mettre en place pour garantir la pérennité de l’état qualifié de l’infrastructure.
Objectifs
A40
Formation inter-entreprises
DUREE 2 jours
PUBLIC
- Membre de l’Assurance Qualité Infor-
matique
- Personnels des services IS et IT
- Responsable de la sécurité informa-
tique
- DSI
- Responsables Qualification/Validation
D12
B14

VA
LID
AT
ION
/ Q
UA
LIF
ICA
TIO
N
15Pour toute formation intra-entreprise : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
Contenu des rapports de validation. Documen-tation d’exploitation : Instruction d’adminis-tration, d’utilisation, de vérification, ... Notions indispensables lors de toute exploitation d’un système ou utilisation d’un équipement : la gestion des modifications (le change control), la vérification périodique et la revalidation.
EXERCICES DE MISE EN SITUATIONEtude d’une approche de validation pour un système informatisé autour de plusieurs équipe-ments. Un QCM vous est proposé afin d’évaluer les connaissances acquises.
1. IntroductionRappels sur la Validation : Définitions et ob-jectifs de la validation. Position des agences officielles. Référentiels réglementaires et normatifs : Description des exigences et propositions des référentiels français, européens, nord-américains et internationaux : BPF, cGMP, BPL, GLP, ICHQ7A, 21 CFR Part 11, GAMP, Pharmaco-pées (PE, USP, JP), normes ISO-NF, ….Principes de validation et de qualification : Les documents de validation à établir ainsi que les règles de base et les bonnes pratiques documentaires qui permettent d’assurer une traçabilité suffisante de vos réalisations.
2. Validation Master Plan (VMP)Présentation de l’objectif et de l’organisation d’un VMP. Comment obtenir un VMP précis, non ambigu et cohérent, ainsi que la matrice des systèmes qui lui est associée.
3. Plan de ValidationPrésentation de l’objectif, des stratégies de rédaction et des matrices de qualification. Comment définir l’organisation, les responsa-bilités, choisir la démarche et les activités de validation dans un Plan de Validation cohérent et non ambigu pour chacun de vos systèmes.
4. Les étapes de qualificationPrésentation / Rappel sur les étapes de qualifi-cation : - QF : Familiarisation avec la Qualification Four-
nisseur - QF (prouver que les éléments sont conçus et réalisés selon les bonnes pratiques techniques et réglementaires en vigueur).
- QD : Familiarisation avec la Qualification de Développement - QD (prouver que toutes les exigences utilisateurs et réglementaires ont été correctement prises en compte).
- QI : Familiarisation avec la Qualification
d’Installation - QI (prouver la bonne instal-lation de vos systèmes).
- QO : Familiarisation avec la Qualification Opé-rationnelle - QO (prouver le bon fonctionne-ment de vos systèmes à l’aide de tests nomi-naux, défaillants et limites / Worst Case).
- QP : Familiarisation avec la Qualification de Performance - QP (prouver la fiabilité et le ser-vice régulier des systèmes).
5. Stratégies et méthodologiesQuelles stratégies et méthodologies mettre en œuvre en fonction des activités des labora-toires, des types de systèmes ou équipements et de leur domaine d’utilisation ? Comment les justifier ? Différences entre Qualification Tech-nique / Fournisseur et Qualification Complé-mentaire / Validation. Quand valider et quand ne faire que de la métrologie ? 6. Les analyses de risques Quelles analyses de risques mettre en œuvre et comment les justifier ? Présentation des principaux types d’analyses de risques disponibles. 7. Etudes de cas : - Un équipement de laboratoire - Un système de gestion des équipement - Un instrument de mesure - Une fiche de calcul
8. La validation et l’organisationLe minimum requis d’un point de vue organisa-tionnel pour supporter efficacement les valida-tions. Quelle organisation mettre en œuvre pour supporter les validations et pourquoi. Quand et pourquoi remplacer les protocoles de qualifica-tion par des procédures de qualification.
9. Conduite des tests, rapports et exploitationFamiliarisation avec le déroulement des tests,
Ne pouvant être considérés comme des équipements de production, plus spécifiques et de taille plus humaine que les énormes systèmes informatisés actuellement déployés, les systèmes et équipements de laboratoires sont au cœur de cette formation qui aborde les spécificités d’une validation historiquement souvent complétée par des feuilles de calcul.Cette formation fait une place importante à la pratique en ne présentant que l’essentiel d’un point de vue théorique et en intégrant des études de cas et des exercices de mise en situation permettant de comprendre comment passer de la théorie à la pratique sur plusieurs types de systèmes et d’équipements.
Validation des Systèmeset des Equipements de Laboratoire (VSEL)
Programme
● Connaître les exigences réglementaires et normatives applicables à la validation et déterminer les informations du dossier de validation indispensables pour être en conformité avec les exigences réglemen-taires de l’AFSSAPS, de l’EMEA et/ou de la FDA (BPF/GMP, BPL/GLP, BPC/GCP, QSR…).
● Savoir définir une stratégie de validation adaptée aux différents types de système et d’équipement ainsi qu’aux différentes activités des laboratoires (Recherche et formulation, Développement et mise au point, contrôle qualité, analyse clinique).
● Savoir valider des systèmes et des équi-pements de laboratoire de façon prag-matique et en conformité avec les recom-mandations du GAMP.
● Connaître les différences entre Qualifica-tion et Métrologie et les implications de ces différences.
● Connaître les différences entre Qualifica-tion Technique / Fournisseur et Qualifica-tion Complémentaire / Validation.
Objectifs
A50
Formation inter-entreprises
PUBLIC
- Correspondant Assurance Qualité pour
le laboratoire
- Responsable de laboratoire
- Technicien de laboratoire chargé de
validation
- Equipes de Maintenance / Métrologie
pour le laboratoire
- Equipes de Qualification / Validation
pour le laboratoire
PÉRIMÈTRE
• Equipements de laboratoires : Chroma-tographie, Spectrométrie, Détermina-tion Particulaires, Dissolutest...
• Instruments de mesures : Titrateur, pHmètre, Balance, COT/TOC, Potentio-mètre…
• Systèmes de gestion d’équipement de laboratoire : Empower, Chemstation, Turbochrom, MAssLynx…
• Enceintes climatisées : Etuve, Réfrigéra-teur, Congélateur…
• Equipements de microbiologie : Lecteur de microplaque LAL, Compteur particulaire…
• Equipements d’environnement : Hotte, Flux laminaire…
• Fiches de calcul
DUREE 2 jours B9

QUALITÉ / RÉGLEMENTATION

QU
ALI
TE
/ R
EG
LEM
EN
TAT
ION
17Pour toute formation intra-entreprise : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
Depuis quelques années, les instances réglementaires Européennes, Américaines et Japonaises communiquent sur les principes de Management de la Qualité. L’ICH Q10 vient compléter ces communications et renforcer les exigences en termes de Système de Management de la Qualité dans le domaine pharmaceutique. La problématique principale n’étant pas uniquement de savoir ce qu’est un Système de Management de la Qualité mais surtout de savoir comment le mettre en œuvre sans bouleverser complètement un système déjà en place et tout en continuant à respecter les principes des Bonnes Pratiques de Fabrication. D’autres textes et normes peuvent aussi aider à se mettre en conformité de façon pragmatique. Il faut aussi gérer les connaissances (Knowledge Management) pour réussir l’amélioration continue.
1. Les objectifs de l’ICH Q10 par rapport aux cGMPQue peut apporter l’ICH Q10 à une industrie pharmaceutique ? Quelles sont les différences entre les cGMP et l’ICH Q10 ? Les bénéfices. Historique.
2. Les exigences de l’ICH Q10Les nouvelles exigences auxquelles doit ré-pondre un système qualité pharmaceutique tout au long du cycle de vie du produit selon l’ICH Q10. Les quatre piliers du Système Qualité Phar-maceutique : 1) Système de surveillance de la performance procédé / processus et de la qualité produit2) Système d’action corrective et préventive (CAPA)3) Système de gestion des modifications4) Système de revue de la performance pro-cédé / processus et de la qualité produit
3. Les exigences des autres textes et guidesQuels sont les autres textes et guides régle-mentaires Européens, Américains et Japonais à prendre en compte ? Quels sont les exigences de ces autres textes et guides réglementaires ? Quelles sont les normes qui peuvent apporter des solutions pragmatiques ?
4. L’approche processus, la dynamique PDCA et la vision transversale « client – client » en industrie pharmaceutique
Qu’est-ce que l’approche processus ? La no-tion de client en industrie pharmaceutique. Où se situe l’assurance qualité dans le système qualité pharmaceutique ? Comment s’imbrique l’ICH Q10 dans les cGMP ?
5. L’analyse de risque comme outil de dia-gnostique des procédés, processus et de la qualité produitEtablir les décisions à partir de données ré-fléchies et terrain. Etape préliminaire pour la mise en place d’indicateurs et la maîtrise des procédés, des processus et de la qualité produit.
6. La direction et la communicationLes responsabilités de la direction. Les revues de direction. Les responsabilités et les res-sources. Les enjeux de la communication dans l’appropriation du Système Qualité Pharmaceutique.
7. Les étapes pour obtenir un système qualité pharmaceutique cohérent avec les objectifs de l’ICH Q10, des cGMP et de l’entrepriseL’état des lieux. Définition de la mise en place, objectifs, ressources, outils de suivis. La mise en œuvre, formation / information, mise en place du plan d’action. Mettre en place des indicateurs pertinents et efficaces. Pilotage, communication interne, revue et amélioration continue. Le Manuel Qualité. Les Plans Qualité. Les procédures.
8. L’amélioration continue Gestion des connaissances (Knowledge Ma-nagement). Identifier les sources de variation. Analyser les données des indicateurs pour évaluer la maîtrise du système qualité phar-maceutique et mettre en place des actions appropriées pour l’amélioration continue.
9. SynthèseLes points importants à retenir.
EXERCICES DE MISE EN SITUATIONPlusieurs exercices sont proposés pendant la formation pour illustrer les points abordés.
Un QCM vous est proposé afin d’évaluer les connaissances acquises.
ICH Q10 : Mise en œuvre pragmatique
Programme
● Connaître les nouvelles exigences de l’ICH Q10 aux différentes étapes du cycle de vie du produit.
● Connaître les piliers d’un système qualité pharmaceutique selon l’ICH Q10.
● Savoir intégrer les exigences de l’ICH Q10 dans le système qualité existant de manière appropriée et proportionnée à l’entreprise et aux différentes étapes du cycle de vie du produit.
● Savoir prendre en compte et intégrer les exigences des autres textes et guides réglementaires Européens, Américains et Japonais relatifs aux systèmes de
management de la qualité ainsi que les normes existantes.
● Savoir définir un processus et mettre en place un pilotage des performances des processus.
● Savoir utiliser le Knowledge Management et la gestion des risques qualité afin de définir des indicateurs pertinents et efficaces.
● Comprendre l’importance de la sensi-bilisation et de la communication sur le système qualité dans l’amélioration de la qualité produit et l’efficacité du système.
Objectifs
B10
Formation inter-entreprises
NOUVEAU
PUBLIC
- Directeur Qualité
- Responsable Assurance Qualité
- Ingénieur Qualité
- Directeur de Production
- Responsable de Production
- Responsable de Processus / Ligne de
Produit
DUREE 2 jours
B30
B103
Vous souhaitez réaliser une
formation sur l’ICH Q8 ?
Contactez-nous + 212 (0)6 61 13 69 28
www.2ssante.com

18
QU
ALI
TE
/ R
EG
LEM
EN
TAT
ION
Pour tout projet de formation : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
Le respect des bonnes pratiques de fabrication garantit la qualité des médicaments. Cette formation permet d’appréhender dans leur ensemble, les textes réglementaires européens, nord-américains et / ou internationaux applicables à la fabrication, l’importation et l’exportation de médicaments, cliniques ou non.
1. Rappels et définitionsMédicament, Principe Actif Pharmaceutique, PAP), Matière première de PAP, Notions d’Innocuité, de Toxicité, Cycle de vie d’un médicament. 2. Organismes et textesOrganismes et réglementations. Principes réglementaires majeurs. Généralités et reconnaissances mutuelles. Présentation des différents textes. Les inspections.
3. Obligations réglementairesPrincipe de l’assurance qualité en milieu pharmaceutique. Différentes approches : ISO, BPF, FDA. Formation. Etudes de « Warning Letters ».
4. Approche détaillée des différents items
4.1 - Management de la qualité Dispositions générales. Personne Qualifiée. Cas particulier des phases cliniques. Cas particulier des intermédiaires.
4.2 - Exigences relatives au personnelQualification du personnel. Formation, expé-rience, suivi et évaluation long terme.
4.3 - Exigences relatives aux locaux et aux équipements Eviter les risques de confusion et toutes sortes de contaminations. Conception et construction.Entretien et nettoyage.4.4 - Exigences relatives aux outils de pro-ductionSur les composants. En cours de fabrication. A l’étiquetage. Au conditionnement.
4.5 - Exigences relatives à la distribution et au stockageIntermédiaires et produits finis. Stockage.Entreposage et distribution.
4.6 - Exigences relatives au contrôle de qualitéLaboratoire de contrôleÉchantillonnage. Tests et libération pour la vente. Etude de stabilité. Echantillothéque.
4.7 - Exigences légales en termes de docu-mentationRecommandations générales.Nettoyage des équipements et livre de bord. Rapports et enregistrements.Enregistrements de production et des contrôles. Dossier de réclamation.Dossier de fabrication (lot).Dossier de conditionnement (lot).Procédures et modes opératoires.
4.8 - Exigences relatives au retour et au rappel de produitRetour de produits pharmaceutiques.
4.9 - Exigences relatives aux réclamationsRéclamations et Rappels de Médicaments.
4.10 - Exigences relatives aux systèmes informatiques21 CFR part 11, Annexe 11.
4.11 - La maîtrise de l’environnementLa métrologieHygiène et microbiologie : les contaminants et les contaminations, hygiène et propreté, les salles propres, le nettoyage. Le contrôle microbiologique. La maintenance.
4.12 - Les auditsAudits externes. Audits internes et auto-inspections.
5. Les évolutions récentes en termes de textes et d’interprétation
6. Les futures évolutions et les textes en préparation
Un QCM vous est proposé afin d’évaluer les connaissances acquises.
BPF et cGMP : principes généraux, retours d’expérience et nouveautés
Programme
● Connaître les organismes chargés de véri-fier l’application des BPF (AFSSAPS, EMEA) et des cGMP (FDA). En comprendre les rôles et les fonctionnements.
● Comprendre le rôle des différents dépar-tements industriels (production, contrôle qualité, R & D, assurance qualité) dans l’ap-plication des principes réglementaires.
● Comprendre les attentes des organismes officiels assurant les inspections et être capable de se préparer à ces inspections.
● Acquérir les connaissances réglemen-taires permettant d’assurer la qualité du produit.
● Connaître les évolutions récentes en termes de textes et d’interprétation.
● Connaître les futures évolutions et les textes en préparation.
Objectifs
B21FORMATION ACTUALISEE
Formation inter-entreprises
PUBLIC
Toute personne devant appliquer ou faire
respecter les BPF et cGMP
DUREE 2 jours
B10
A15
A131
D26
B27
D7
B104
Toutes nos formations sont
disponibles en anglais
Contactez-nous + 212 (0)6 61 13 69 28
www.2ssante.com

QU
ALI
TE
/ R
EG
LEM
EN
TAT
ION
19Pour toute formation intra-entreprise : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
Le sommaire standard de chaque formation est disponible sur simple demande.
Si vous le souhaitez, nous pouvons adapter le contenu de ces formations à votre contexte.
Notre démarche pour cette adaptation à vos besoins consiste à : - évaluer le niveau des participants sur les
thèmes abordés, - recenser les attentes des participants et de
leurs managers, - recenser les procédures en place dans l’en-
treprise et les intégrer à la formation, - tenir compte de la politique qualité de
l’entreprise et du système qualité en place.
Ces formations « terrain » sont construites de façon ludique et agrémentées de petits jeux (chercher les erreurs, questions / réponses …) afin de faciliter l’apprentissage.
Evaluation des connaissances acquises et conclusion du séminaire. Un QCM vous est proposé afin d’évaluer les connaissances acquises. Vérification de l’atteinte des objectifs établis en début de séminaire.
A partir de vos propres cas concrets, CVO-CyberConseil vous apporte de nombreuses solutions grâce à un transfert de connais-sances théoriques et une mise en application pratique. Ces formations-actions d’une durée à définir sont proposées à partir de différents thèmes.
Formations disponibles sur les thèmes suivants :
B21A Présentation des réglementations cGMPs / BPF
B21B Organisation, personnel et cGMPs / BPF
B21C Procédures, audit et cGMPs / BPF
B21D Réception des matières et cGMPs / BPF
B21E Echantillonnage et cGMPs / BPF
B21F Manipulation, utilisation des matières et cGMPs/BPF
B21G Bâtiments, locaux et cGMPs / BPF
B21H Equipements et cGMPs / BPF
B21I Métrologie, étalonnage et cGMPs / BPF
B21J Processus de validation et cGMPs / BPF
B21K Documentation de validation et cGMPs / BPF
B21L Validation de l’informatique et cGMPs / BPF
B21M Assurance qualité et cGMPs / BPF
B21N Contrôle qualité et cGMPs / BPF
B21O Documentation et cGMPs / BPF
B21P Système de quarantaine et cGMPs / BPF
Formation Action des opérateurs sur siteaux BPF et cGMP
Permettre aux acteurs du secteur des Industries de la Santé de :
● Connaître les exigences réglementaires
européennes et américaines.
● Se préparer à une inspection et à un audit.
● Comprendre les normes réglementaires par
rapport aux activités de leurs départements
ou services.
Objectifs
B211
PUBLIC
- Assurance Qualité
- Production
DUREE De deux heures à une journée selon vos besoins.
Toutes nos formations sont
réalisables en intra-entreprise
Contactez-nous + 212 (0)6 61 13 69 28
www.2ssante.com

20
QU
ALI
TE
/ R
EG
LEM
EN
TAT
ION
Pour tout projet de formation : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
L’ ISO 22716 définit les règles permettant de garantir que les produits sont fabriqués, conditionnés, contrôlés et stockés selon les mesures adaptées à leur utilisation et leur spécificité. Tout le personnel doit avoir une connaissance de base de tous les éléments pouvant affecter les BPF et par conséquent la qualité des produits.
1. Principes et objectifs des BPF pour les produits cosmétiquesEvolution de la réglementation. La norme internationale ISO 22716 : Historique, archi-tecture du texte. ISO 22716 et les BPF phar-maceutiques. Le projet de simplification de la législation européenne concernant les produits cosmétiques. Quoi de neuf dans la législation américaine ?
2. Personnel et FormationQuels sont les points importants dans l’orga-nisation : Organigramme, effectif, personnel qualifié ? Importance de la formation pour la conformité GMP : les requis de formation et le plan de formation. Comment documenter les formations et réaliser le suivi ? Pourquoi et comment réaliser les évaluations des for-mations ?
3. Hygiène et Santé du personnelComment éviter la contamination des pro-duits ? Quels sont les dispositions à prendre pour l’hygiène et la santé du personnel ? Pourquoi mettre en place un Programme d’Hygiène ?
4. Les locauxComprendre les exigences de conception générale et la ségrégation des zones. Com-prendre les exigences des flux matériel, per-sonnel et produits et leur rôle dans la préven-tion de la Contamination Croisée. Comment démontrer l’efficacité des agents de net-toyage ? Définir le programme de nettoyage – désinfection. Mettre en place les moyens de protection contre les parasites. Comment gérer les déchets ? Types de déchets, flux, contenants, destruction.
5. L’Assurance Qualité et le Contrôle Qualité
Quels sont les Rôles et Responsabilités de l’Assurance Qualité ? Quels sont les Rôles et Responsabilités de l’unité de Contrôle Qua-lité ? Le besoin d’indépendance par rapport aux autres unités de l’usine. Assurance Quali-té vs. Control Qualité. Qu’est ce que le SISPQ ?
6. Les équipementsPourquoi définir des spécifications de concep-tion pour les équipements, les matériaux utilisés et les consommables ? Comprendre les exigences pour le nettoyage et l’intérêt du Programme de Maintenance Préventive. Calibrer/Etalonner : Quoi et Comment ? Sys-tème de remplacement.
7. Matières premières et les articles de conditionnementComprendre les exigences de Spécifications pour les matières premières et les articles de conditionnement. La qualification des four-nisseurs : Quoi et Comment ? Comprendre les règles à appliquer à la réception et au stockage des matières. Comment identifier et assurer la traçabilité des produits ? Pour-quoi réévaluer les matières premières ? Com-prendre les exigences de qualité pour l’eau utilisée en production. Comment traiter les produits hors spécification ?
8. La production et le contrôle du processusComprendre les exigences concernant les activités de préfabrication et les opérations de fabrication. Quels sont les exigences concernant les activités de conditionnement et l’étiquetage du produit ? Quels aspects des GMP appliqués à votre travail quotidien ? Pourquoi des critères d’acceptation ? Mise en quarantaine, libération des produits et leur stockage. Comment traiter les produits hors spécification ? Expédition.
9. Le laboratoire de Contrôle de la QualitéComment assurer la fiabilité et la qualité des résultats au laboratoire ? Quels sont les requis de l’échantillonnage ? Pourquoi définir des critères d’acceptation ? Comment gérer les résultats hors spécification ? Qu’est ce qu’un échantillon témoin ?
10. La sous-traitanceQuels sont exigences concernant la sous-traitance des activités ? Pourquoi et comment évaluer le fournisseur ?
11. Les déviations et la gestion des modi-ficationsPourquoi est-il important de documenter les Déviations ? La règle qualité des 1/10/100. Comment assurer la qualité du produit par la maitrise des changements ?
12. La gestion des Réclamations et le Rap-pelComment gérer les réclamations ? Com-prendre les exigences concernant le rappel de lot. Comment prévenir la récurrence des défauts ?
13. Audit Interne et inspectionComprendre l’importance de l’audit interne dans la surveillance de l’application des BPF. Comment gérer et suivre l’audit interne ? Quelles sont les modalités d’une inspection cosmétique ? Comment préparer l’audit avec succès ? Que recherchent les inspecteurs ?
14. La documentationArchitecture et gestion du système documen-taire. Quelles sont les procédures requises par les BPF ? Enregistrements et rapports : qu’est-ce qu’un enregistrement ? Comprendre les bonnes pratiques de documentation. Archivage : Quoi et Comment ?
EXERCICES DE MISE EN SITUATIONPlusieurs exercices sont proposés pendant la formation pour illustrer les points abordés. Un QCM est proposé afin d’évaluer les connaissances acquises.
BPF Cosmétiques (ISO 22716) : principes et objectifs
Programme
● Comprendre les principes fondamentaux et l’esprit des BPF pour les mettre en œuvre.
● Acquérir les connaissances réglemen-
taires afin d’assurer la qualité du produit.
● Connaître les exigences et les caracté-ristiques de la norme ISO 22716 pour
la fabrication, le conditionnement, le contrôle et le stockage du produit.
● Comprendre les rôles des unités de Contrôle Qualité et d’Assurance Qualité.
Objectifs
B220
Formation inter-entreprises
FORMATION ACTUALISEE
PUBLIC
Cette formation est destinée aux
personnes impliquées dans toutes les
phases de fabrication des produits
cosmétiques :
- Assurance et Contrôle Qualité
- Validation
- Fabrication
- Maintenance
- Sous-traitants de la Cosmétique
DUREE 2 jours
A15
B10
B104
D26

QU
ALI
TE
/ R
EG
LEM
EN
TAT
ION
21Pour toute formation intra-entreprise : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
BPL, GLP, 21CFR58, Toxicologie, Sécurité des produits, AMD – Acceptation Mutuelle des DonnéesL’objectif premier des Principes de l’OCDE relatifs aux BPL est d’assurer l’obtention de données d’essai fiables et de grande qualité sur la sécurité des substances et préparations chimiques industrielles, dans le cadre de l’harmonisation des procédures d’essai aux fins de l’acceptation mutuelle des données (AMD).
Bonnes pratiques de laboratoires pour les essais pré-cliniques : conformité, AQ et audit
Objectifs
● Comprendre les principes de gestion des BLP, OCDE/GLP, 21CFR58 pour les installa-tions engagées dans les études de toxico-logie et de sécurité des produits.
● Appréhender les exigences pour l’organi-sation des installations d’essai et les condi-tions dans lesquelles doivent être exécutées les études de sécurité précliniques. Les cas particuliers de la sous-traitance et de la conduite de l’étude multi-site seront pré-sentés aussi.
● Comprendre les fonctions et responsabi-lités du Directeur d’Etude, de l’Investiga-teur Principal et de l’Assurance Qualité.
● Savoir l’essentiel pour la mise en place d’un système d’archivage et pour la conformité de vos systèmes informatisés selon les BPL.
● Revenir sur la nécessité d’évaluer la conformité de l’installation aux BPL / GLP.
Programme
1. Introduction et Contexte RèglementaireHistoire des BPL/GLP (OCDE, FDA). Harmonisa-tion technique (CIH). A qui s’adressent les BPL et à qui ne s’adressent-elles pas ? Quels sont les objectifs des BPL ? Quels sont les fondements et grands principes BPL/GLP ? Définitions.
2. Organisation et PersonnelRôles et Responsabilités des acteurs de l’étude : Management de l’installation d’essai, Directeur d’Etude, Responsable Principal des essais et du Personnel de l’Etude.
3. Assurance Qualité et BPLLes Responsabilités de l’unité d’AQ. Qualifica-tions du personnel. Rôle de l’assurance qualité dans l’élaboration des modes opératoires nor-malisés et des plans d’étude. Gestion des procé-dures, Modes Opératoires Normalisés requises par les BPL. Inspections AQ sur les études, sur les installations et sur les procédés. Rapports d’inspection. Audits des données et du rapport final. Déclaration sur le programme d’assurance qualité.
4. Installations d’essais et EquipementsConception des locaux pour le système d’essai et les activités de l’étude. Isolation des projets. Ségrégation des zones. Salle d’Archive : Stoc-kage des données et des spécimens et le contrôle des accès. Collecte, Stockage et Eva-cuation des déchets. Nettoyage et Déconta-mination. Conformité des Equipements : spé-cifications, calibration, qualification, validation, maintenance, nettoyage. Disqualification de l’installation.
5. Système et éléments d’essai et de réfé-renceRequis pour les systèmes d’essai physiques et chimiques. Requis pour les systèmes d’essais biologiques. Réception, manutention, échan-
tillonnage et stockage. Caractérisation de l’élément d’essai : identification, stabilité et traçabilité.
6. Réalisation de l’Etude Protocole et identification de l’étude. Données collectées pendant l’Etude. Gestion des amen-dements et déviations. Rapport d’Etude. Archi-vage. Les BPL et les tests courts.
7. Application des BPL à l’archivageLes locaux d’archives : conditions d’archivage, Reprise après sinistre. Sécurité : physique et opérationnelle, Accès aux archives. Procédures d’Archivage : Modes opératoires normalisés. Pièces et matériaux à conserver. Index de l’ar-chivage. Période de conservation et destruction. Placement de pièces et de matériaux dans les archives, Transferts. Elimination de pièces et de matériaux. L’archivage de dossiers électro-niques : décision de conserver des pièces sous forme électronique, Supports de stockage, Zone d’archivage définie à l’intérieur d’un système informatique, Système d’archivage électronique dédié, Entretien et préservation des dossiers électroniques.Assurance qua-lité. Dépôts d’archives sous-traitants. Mesures à prendre suite à la fermeture d’archives.
8. Application des BPL à l’organisation et la conduite des études multi-sitesCommunication. Attributions respectives : don-neur d’ordre, direction de l’installation d’essai, direction du site d’essai, le Directeur de l’étude, Responsable principal, Personnel travaillant à l’étude. AQ des études multi-sites : Attribu-tions du coordonnateur de l’AQ, Attributions de l’unité AQ du site d’essai. Le schéma direc-teur de l’ensemble des sites intéressés. Le plan unique de l’étude. Réalisation de l’étude : instal-lations, équipements, contrôle et transparence des matériaux d’étude. Les comptes rendu des
résultats de l’étude. Les modes opératoires nor-malisés. Stockage des relevés et matériaux.
9. Travailler avec des fournisseurs et des ORCsEvaluation et sélection du laboratoire sous-traitant. Monitoring de l’étude : rôle et res-ponsabilités du moniteur, ses méthodes, son organisation et la planification de son travail, sa capacité à identifier et résoudre les problèmes, ses qualités scientifiques et relationnelles. La décision de sous-traiter une étude. Critères de choix et sélections d’un laboratoire sous-traitant : l’évaluation préalable. Mise en place de l’étude sous-traitée. Démarrage, suivi et clôture de l’étude. Les points clés lors d’une visite. Conseils pratiques pour un monitoring efficace.
10. Application des principes BPL aux sys-tèmes informatisésResponsabilités. Formation. Installations et équipements. Maintenance et reprise après un sinistre. Données critiques BPL. Sécurité phy-sique, logique et intégrité des données. Validation des systèmes informatisés. Documentation. Sauvegarde. Archives.
11. Inspections RéglementairesQue recherchent les inspecteurs ? Comment s’y préparer ? Les systèmes d’inspection et de véri-fication de la mise en conformité aux BPL.
EXERCICES DE MISE EN SITUATIONPlusieurs exercices sont proposés pendant la formation pour illustrer les points abordés.Un QCM vous est proposé afin d’évaluer les connaissances acquises.
B25
Formation inter-entreprises
FORMATION ACTUALISEE
PUBLIC
Cette formation s’adresse à tout per-
sonnel des industries, ORC ou centre de
recherche, spécialement :
- Moniteur
- Directeur d’Etude
- Personnel de l’Etude
- Responsable Principal des essais
- Assurance Qualité (AQ)
- Auditeurs
- Attaché de Recherche Clinique (ARC)
DUREE 2 jours
B10
D22
B104
B27

22
QU
ALI
TE
/ R
EG
LEM
EN
TAT
ION
Pour tout projet de formation : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
Testez GxP KR Santé/Pharma gratuitement !
e-mail :
www.gxpmanager.com
10. Application des BCP aux systèmes in-formatisésExigences des BPC/GCP. Requis pour les données source PIC/S, CEDISC. La conserva-tion électronique des dossiers source. Impact sur les investigateurs/chercheurs. Principes généraux de Validation. Principales difficultés de la validation sur site d’investigation, en particulier les centres de santé. Cas du Dossier Médical Electronique, eCRF.
EXERCICES DE MISE EN SITUATIONPlusieurs exercices sont proposés pendant la formation pour illustrer les points abordés.
Un QCM vous est proposé afin d’évaluer les connaissances acquises.
BPC, GLP, essais cliniques, protection de la vie privée, comités d’éthique La mise en œuvre d’essais cliniques implique de suivre des règles très strictes dans plusieurs domaines. Les responsabilités sont définies clairement pour chaque domaine et pour chaque activité et elles doivent être strictement respectées. Pour s’assurer que l’on ne se met pas en porte à faux par rapport à toutes ces exigences, il est important de bien comprendre les principes fondateurs de l’éthique dans les essais cliniques ainsi que toutes les réglementations associées aux essais.
Bonnes pratiques cliniques pour les essais de médicament : éthique et qualité des données
Programme
● Comprendre les responsabilités spé-cifiques et les obligations de tous les individus impliqués dans la recherche clinique pour de Nouvelles Applications de Médicament, en particulier celles des Investigateurs, Promoteur et Moniteurs.
● Connaître les principes fondateurs de l’éthique dans les essais cliniques, les rôles et responsabilités des comités d’éthique ainsi que la règlementation de la protec-tion de la vie privée.
● Comprendre les exigences concernant le produit de recherche et le placebo.
● Savoir mettre en place une surveillance de l’essai et procéder à une vérification en cours d’étude.
● Comprendre les modalités d’assurance qualité et de contrôle qualité afin que les essais soient réalisés conformément au protocole et aux BPC.
● Comprendre les exigences concernant les données de l’étude.
Objectifs
B26
Formation inter-entreprises
FORMATION ACTUALISEE
PUBLIC
Cette formation est destinée aux
personnes travaillant dans les industries
pharmaceutiques et biopharmaceutiques
et impliquées dans toutes les phases de
la recherche clinique :
- Moniteur
- Attaché de Recherche Clinique, ARC
- Coordinateur
- Investigateur/Chercheur
- Affaires Réglementaires
- Communication médicale
- Organisme de recherche sous contrat
(ORC)
DUREE 2 jours 1. Introduction et contexte règlementaireProcessus de développement du médica-ment. Historique des Bonnes Pratiques Cli-nique : Code de Nuremberg, Déclaration d’Helsinki, CIH, OMS, CFR, Harmonisation en Europe, A qui s’adressent les BPC ? Quels sont les acteurs principaux de l’étude clinique ? Principes inhérents des BPC : Protection des sujets, Qualité des données, Transparence dans la conduite de l’étude.
2. Pré-requis à l’Etude Clinique Justification de l’étude. Conception de l’étude-: sécurité, efficacité, principaux résultats, essais en aveugle ou ouvert, randomisation, taille de l’étude, Statistiques et Calculs. Les données supportant le médicament destiné à l’essai, L’Investigateur et les sites d’investigation, Essais multicentriques, Autorisation pour démarrer l’étude.
3. Partage des responsabilités (promoteur, comité d’éthique, investigateur)Responsabilités de l’investigateur/chercheur : qualification, délégation, conformité au proto-cole et requis BPC, protection sujets, consen-tement éclairé, gestion des enregistrements de l’étude, soin médical, ressources, comités. Responsabilités du moniteur : Qualifications, évaluation du site d’investigation, Surveillance, Vérification, Formation du personnel, CRF, gestion des données de l’étude, Notification, Non conformités, Rapport de surveillance. Responsabilité du Comité d’éthique indépen-dant (CEI) et du Comité d’examen de l’établis-sement (CEE) : responsabilités, composition, fonction et activités, procédures, dossier.
4. Protection des sujets participant à l’essai Déclaration d’Helsinki. Comité d’éthique. Consentement éclairé du sujet. Confidenti-alité.
5. Surveillance Gestion et enregistrements des effets indési-rables. Incidents thérapeutiques graves (ITG).Suivi et résolution. Suivi et tendance. Rappor-ter les effets indésirables.
6. La documentation de l’étudeDocuments source. Requis du CDISC. Le pro-tocole d’Etude et les modifications. Déviations par rapport au protocole. La Brochure de l’in-vestigateur. Consentement éclairé. Rapport de surveillance. Notification. Liste des codes d’identification. Registre des échantillons. Rapport provisoire. Rapport final.
7. Assurance Qualité et Conduite de l’Etude CliniqueProcédures. Mode Opératoire Normalisé. Vé-rification des données sources. Formation. Signatures. Méthodes pour les laboratoires. Manipulation et Sauvegarde des données de l’Etude Clinique : responsabilités, accès aux données, archivage des données.
8. Produit de RechercheApprovisionnement. Stockage. Etiquetage.Conditionnement. Manipulation et Respon-sabilité pour le produit d’essai.
9. Inspection Autorités de réglementation du médicament-: rôles et responsabilités, Inspections sur site.Inspection des Essais Cliniques.
D26
B10
B27

23
A qui s’adresse GXP KR SANTÉ / PHARMA ? Cette solution s’adresse à tous les acteurs des entreprises de la Santé qui ont besoin de connaître immédiatement l’ensemble des exigences réglementaires à jour pour prendre les bonnes décisions.
GXP KR Santé/Pharma répond aux attentes de l’ensemble des personnes qui intervien-nent au niveau des Affaires Réglementaires, de la Qualité, de la Validation, de l’Ingénierie et de l’Informatique qui doivent en perma-nence réaliser des projets ou des arbitrages conformes aux exigences réglementaires.
Exemples d’utilisation : - intégrer les exigences réglementaires spécifiques dans une procédure opéra-tionnelle,
- préparer un audit fournisseur ou interne selon un référentiel spécifique (BPL ou ICH, ...),
- analyser les textes réglementaires ap-plicables pour un nouveau projet et les utiliser directement comme exigences particulières,
- assurer la traçabilité complète entre les exigences réglementaires et les proto-coles de qualification.
GXP KR Santé/Pharma est
un accès Web aux règle-
mentations et aux standards
avec plus de 1000 textes
internationaux.
C’est le moyen le plus facile
d’accéder aux normes et
règlementations dans les
secteurs pharmaceutique,
dispositifs médicaux,
cosmétique, principes actifs.
GXP KR Santé/Pharma est très
simple d’utilisation grâce à
ses fonctionnalités avancées
telles qu’un puissant moteur
de recherche, la gestion des
favoris, les alertes e-mail,
l’assistance aux recherches et
plus encore.
GXP KR Santé/Pharma est une solution économique qui vous fera gagner de lon-gues heures de recherche et vous permet-tra d’obtenir instantanément les réponses fiables à vos questions. Des remises par quantité sur les abonnements permettent
de faire bénéficier tous les acteurs concer-nés de l’entreprise d’une base de données règlementaires facilement accessible et à jour.
Une offre économique
GXP KR Santé/Pharma met à votre dispo-sition la réglementation en vigueur : les directives, les lois, les arrêtés et les préco-nisations des instances gouvernementales (guides, guidances, guidelines, présenta-tions) en matière de dispositifs médicaux,
médicaments à usage humain ou vétéri-naire, produits cosmétiques, etc. Retrouvez les informations émanant de plus de 30 organismes émetteurs nationaux et in-ternationaux tels que FDA, AFSSAPS, ICH, EMEA, ...
Une base de données de textes réglementaires applicables aux Industries de la Santé
Testez GxP KR Santé/Pharma gratuitement !
e-mail :
www.gxpmanager.com
GXP KR Santé/Pharma : la base web de données règlementairesdes Industries de la Santé
Une solution pour les Industries de la Santé
GxP KRSANTE
PHARMA
Téléphone : +33 (0)4 26 100 810E-mail : [email protected]
www.gxpmanager.com
►Un puissant moteur de recherche
►Gestion des favoris
►Alertes e-mail
►Assistance aux recherches
►Recherche « full text »

24
QU
ALI
TE
/ R
EG
LEM
EN
TAT
ION
Pour tout projet de formation : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
1. Enjeu et domaine d’applicationPanorama des textes majeurs sur la signature électronique. Définition d’un enregistrement et d’une signature électronique. Les objectifs de la FDA à l’origine du 21 CFR part 11. Les objectifs de l’annexe 11 des GMP Europe. Les objectifs du PI011-3 du PIC/S. Déterminer par-mi ses propres systèmes, ceux qui sont soumis aux exigences relatives aux enregistrements et signatures électroniques. Identifier les enregis-trements électroniques. Dans quelle mesure des signatures électroniques peuvent-elles/doivent-elles être mises en place ?
2. 21 CFR part 11 - Exigences sur les enregis-trements électroniquesDistinction entre un « système ouvert » et un « système fermé ». Conséquences orga-nisationnelles, juridiques et techniques de cette distinction. Exigences du 21 CFR part 11 concernant les enregistrements électroniques (Electronic Records) : validation, documenta-tion du système, maîtrise des modifications, gestion des accès, audit trail, ... Qu’est ce que le guide «Scope and Application» a-t-il changé ? La notion d’analyse de risques est intégrée partout !
3. 21 CFR part 11 - Exigences sur les signa-tures électroniquesPanorama des signatures et de leurs caracté-ristiques : Signatures manuscrites, identifiants, mots de passe, biométrie, cartes à puce… Exigences du 21 CFR part 11 selon les types de signature utilisés : manifestation de la signa-ture, lien signature-enregistrement, exigences
sur les mots de passe, déclaration à la FDA, …
4. GMP Europe Annexe 11 - Exigences sur les enregistrements électroniquesExigences de l’annexe 11 concernant les enregistrements électroniques : validation, documentation du système, maîtrise des modifications, gestion des accès, piste pour audit, …. Connaître les différences entre 21 CFR Part 11 et Annexe 11.
5. PIC/S PI011-3 - Recommandations sur les enregistrements électroniquesRecommandations du guide PIC/S PI011-3 concernant les enregistrements électroniques. Connaître les implications du PIC/S PI011-3.
6. Position de la FDALe débat lancé par l’industrie. L’attitude de la FDA vis-à-vis des systèmes existants (“Legacy systems”). Revue des 483 et des Warning Letters publiés sur les thèmes du 21 CFR part 11 de-puis 1997. Réflexion sur les systèmes hybrides. La nécessité du plan de mise en conformité. Les dernières évolutions de l’interprétation du texte.
7. La démarche de mise en conformitéMise en place d’une structure projet pour la mise en conformité ERES. Mise en place d’un questionnaire. Inventaire des systèmes et véri-fication de conformité au référentiel. Mesures immédiates à prendre quand un système n’est pas conforme et présente des risques. Mise en place des procédures. Ordre de grandeur des budgets et délais nécessaires.
Les systèmes utilisant des enregistrements et des signatures électroniques doivent être en conformité vis-à-vis du texte 21 CFR Part 11 en fonction des exigences définies et ce, dès leur conception ou en rétrospectif tout en prenant en compte les évolutions de la position de la FDA depuis la parution du texte en 1997. Toutefois ce n’est pas le seul texte existant et au niveau Européen il y a aussi l’annexe 11 des GMP et le document du PIC/S PI011-3 à prendre en compte. D’autre part, tout ceci est en pleine évolution et il est important d’essayer d’envisager ce que sera l’avenir dans ce domaine afin de ne pas concevoir des systèmes qui risqueraient d’être non-conformes avant d’être utilisés. La nouvelle annexe 11 des GMP qui est en revue et le nouveau 21 CFR Part 11 devraient être disponibles fin 2009. Que vont-ils changer ? Le PIC/S n’est plus seulement Européen mais la FDA et d’autres instances réglementaires y contribuent et il devient la référence comme pour la nouvelle version du GAMP par exemple, on va donc de plus en plus vers un vrai consensus !
8. Cahier des charges et validation des nou-veaux systèmesDéfinition (dans un cahier des charges) et évaluation (dans le cadre d’un appel d’offres) des exigences de conformité concernant les enregistrements électroniques et les signa-tures électroniques. Adaptation du discours au niveau de maturité. Les principaux écueils tech-nologiques (sécurité, audit trail, horodatage, archivage, ...). Les principes de la validation des systèmes soumis aux exigences ERES.
9. Les évolutions futuresQue sera le nouveau 21 CFR part 11 ? L’annexe 11 des GMP est en cours de revue, mais est-ce que ce sera une évolution ou une révolution ? Est-ce que le PI011-3 du PIC/S va devenir la référence mondiale ?
EXERCICES DE MISE EN SITUATIONPlusieurs exercices sont proposés pendant la formation pour illustrer les points abordés.Un QCM vous est proposé afin d’évaluer les connaissances acquises.
Enregistrements Electroniques et Gestion des Systèmes Informatisés, il y a un consensus !
Programme
● Comprendre les enjeux du 21 CFR part 11, son domaine d’application et ses exigences.
● Comprendre les exigences de l’annexe 11 des GMP Europe (LDP 11des BPF France).
● Connaître les dernières évolutions des exigences Européennes et Américaines.
● Savoir évaluer la conformité des systèmes existants.
● Savoir planifier et mettre en œuvre la démarche de mise en conformité.
● Etre capable de spécifier et d’évaluer la conformité au 21 CFR part 11 lors de l’acquisition d’un nouveau système.
● Connaître dans quel sens ont et vont continuer d’évoluer les règlementations liées aux enregistrements électroniques avec la prise en compte de l’ISO 17799 (Gestion de la sécurité des informations) et du PI011-3 du PIC/S (Bonnes pratiques pour les systèmes informatisés dans des environnements GxP).
Objectifs
B27
Formation inter-entreprises
FORMATION ACTUALISEE
DUREE 2 jours
PUBLIC
- Responsable Validation/Qualification
- Responsables Informatiques (Projets et
Exploitation)
- Responsables d’Ingénierie (Projets et
Exploitation)
- Responsables Qualité
- Chefs de Projet des systèmes
automatisés et informatisés
B8
C20

QU
ALI
TE
/ R
EG
LEM
EN
TAT
ION
25Pour toute formation intra-entreprise : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
La commercialisation et la production des différentes classes de dispositifs médicaux doivent être réalisées dans le respect des réglementations européennes et/ou américaines et des textes normatifs de type ISO.
1. Les enjeux des réglementations appli-cables aux Dispositifs MédicauxRisques liés aux Dispositifs Médicaux (risques
client / risques fournisseur). Présentation des
environnements réglementaires Européen
(CE) et Américain. Différences principales
entre les deux systèmes.
2. Réglementation liée au marquage CE des Dispositifs MédicauxChamps respectifs d’application et principes
des 3 directives européennes. Principes
de classification. Notion de Marquage CE.
Limites et zones d’ombre des directives euro-
péennes. Le système européen de vigilance-:
l’organisation, les responsabilités des fabri-
cants et des distributeurs. Mise en œuvre des
directives : les rôles des fabricants et des
organismes notifiés, la chronologie de mise
en œuvre, la documentation technique.
3. Evaluation de la conformité CELes différentes démarches de conformité CE-:
de la déclaration CE à la mise en place d’un
système complet d’assurance qualité. Impacts
en termes d’évaluation. Choix de la démarche
la mieux adaptée.
4. Détails de l’ISO 13485 :2003Domaine d’application de la réglementation.
Périmètre, responsabilités, système Assu-
rance Qualité et son système de suivi.
Exigences liées aux activités de conception,
validation, production, achats, et contrôle
Qualité.
Exigences documentaires.
Exigences liées aux activités de packaging,
manutention, stockage, distribution, SAV et
suivi des réclamations client. Exigences liées
à la documentation technique.
5. Environnement réglementaire FDA : introduction et classificationHistorique de la réglementation américaine.
Principes de la classification FDA. Différences
avec les classifications CE. Contrôles appli-
cables selon la classification. Gestion des
modifications de classification : pourquoi ?
Comment ?
6. Documentation nécessaire à l’exporta-tion vers les USAEnregistrement de l’établissement et ins-
cription des dispositifs. Déclaration avant
mise sur le marché : le 510(k). Contenu des
documents de commercialisation : PreMarket
Approval (PMA), dossier de données cliniques,
documents d’accompagnement et de tra-
cking.
7. La réglementation 21 CFR 8xx en général, et 21 CFR 820 en particulierDomaine d’application de la réglementation.
Périmètre, responsabilités, système Assurance
Qualité et son système de suivi.
Mise en œuvre d’un outil d’amélioration
continu (le feedback). Exigences liées aux
activités de conception, validation, produc-
tion, achats, et contrôle Qualité. Exigences
documentaires. Dossiers FDA : Design His-
tory File (DHF), Device Master Record (DMR),
Device History Record (DHR), Quality System
Record (QSR).
Exigences liées aux activités de packaging,
manutention, stockage, distribution, SAV et
suivi des réclamations client. Exigences liées
à la documentation technique.
Dossier FDA de matériovigilance (MDR).
8. Savoir aborder une inspection FDA
9. Ecarts entre les directives CE, les régle-mentations FDA, l’ISO 13485:2003 et l‘ISO 9001:2008Principales différences entre 21 CFR 820, ISO
9001:2008 et l’ISO 13485:2003. Ecarts à com-
bler pour une entreprise détentrice d’un mar-
quage CE désireuse d’élargir son marché aux
Etats-Unis.
Un QCM vous est proposé afin d’évaluer les
connaissances acquises.
Réglementation des dispositifs médicaux : USA FDA, Europe MDD et marquage CE
● Connaître et comprendre les enviro-
nnements réglementaires (21 CFR 8xx,
510k, directives 90/385, 93/42, 98/79, ISO
13485:2003).
● Maîtriser les éléments du dossier de mise
sur le marché pour les USA.
● Comprendre les principes d’évaluation de
la conformité CE et la structure documen-
taire associée.
● Connaître les systèmes américains et euro-
péens de vigilance.
Objectifs
Programme
B28FORMATION ACTUALISEE
PUBLIC
- Assurance Qualité
- Informatique
- Ingénierie et automatisme
- Affaires Règlementaires
B29
Pour développer vos compétences, nous vous conseillons également :
B8

26
QU
ALI
TE
/ R
EG
LEM
EN
TAT
ION
Pour tout projet de formation : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
1. Approche de la réglementationPrésentation des différentes agences et orga-nismes : AFSSAPS, EMEA, FDA, MHW, ISO, ICH, … Les accords de reconnaissance mutuelle.
2. Introduction à l’ICH Q7L’ICH Q7 est il opposable ? Présentation du contenu de l’ICH Q7. Comparaison ICH Q7 / 21 CFR parts 210 et 211. Comment cibler les priorités pour répondre aux exigences de l’ICH Q7 ?
3. Principes de l’assurance qualitéImportance des audits internes et des revues qualité produit.
4. PersonnelImportance de la formation et sa documen-tation.
5. Locaux et équipements Pour répondre aux exigences de cette partie, il faut commencer par analyser les risques liés à l’environnement du produit. Prévenir les mélanges et la contamination. Ne pas oublier que le meilleur vecteur de contamination, c’est l’homme. Importance d’un habillage adapté et de règles d’habillage strictes. La place de la métrologie et de la maintenance préventive pour les bâtiments, équipements, instruments, utilités.
6. Systèmes automatisés et informatisés Que doit-on appliquer : 21 CFR part 11, Annexe 11, PIC/S PI011 ? Donnée électronique ou do-nnée manuscrite, où est la donnée brute ?
7. Documentation et enregistrementsProcédures, Modes opératoires, Supports d’en-
registrements, Documentation technique-: qu’est ce qu’ils doivent contenir ? Comment doivent-ils être maîtrisés ?
8. Flux matièresEvaluation des fournisseurs de matières cri-tiques. Combien faire de tests pour vérifier l’identification de chaque lot ?Exigences spécifiques à la préparation de charge. Retraitement, Récupération.Quand a-t-on le droit de faire des mélanges ?Conditionnement, étiquetage, stockage et distribution.
9. EchantillonnageRègles de base. Quels sont les risques liés à l’échantillonnage ? Exigences pour l’échan-tillonnage des matières premières, en cours de production, des articles de conditionnement. Echantillons destinés à la stabilité.
10. ValidationLa politique globale de validation de la société. Les pré-requis à une validation de procédé de fabrication ou de nettoyage. Qualification des équipements critiques : QD, QI, QO, QP.
11. Contrôle QualitéAutres textes impliqués : ICH Q1, ICH Q2, ICH Q3, ICH Q6. Validation des méthodes analy-tiques. Critères de validation d’une méthode d’analyse. Réactifs et étalons. La gestion des Hors Spécifications (OOS) et le cas BARR.
12. Maîtrise du changementLe processus de gestion des modifications.
13. Relations clients-fournisseursLa gestion des retours. La gestion des plaintes et des rappels.
L’ICH Q7, reconnu par les principaux pays importateurs et exportateurs de produits pharmaceutiques comme le seul texte applicable pour la fabrication et le commerce de principes actifs pharmaceutiques et d’intermédiaires, se révèle un guide particulièrement riche et détaillé, qui précise et met à jour les attentes aussi bien des législateurs que des clients industriels. C’est aussi le texte de référence imposé par les instances réglementaires américaines et européennes pour les audits des fournisseurs de principes actifs.
14. ACNDRR - Agents, Courtiers, Négo-ciants, Distributeurs, Re-conditionneurs et Ré-étiqueteursA qui s’adresse cette nouvelle section et pourquoi ?
15. Culture cellulaire et fermentation - CCFAutres sources de guide pour la culture cellu-laire et la fermentation : ICH Q5, ICH Q6.Pourquoi y a-t-il une section séparée pour la culture cellulaire / fermentation ? Qu’est ce qui n’est pas couvert par cette section ? Ou commence les GMP pour la culture cellulaire ou la fermentation ? Bioburden et contamina-tion. Mise en valeur des questions CCF venant d’autres sections.
16. API destinés aux essais cliniquesQuelles sont les particularités des lots fabriqués pour réaliser des essais cliniques ?
17. Les inspectionsComparaison des approches FDA et Euro-péennes. Constat sur les pratiques actuelles des industriels autour de l’application de l’ICH Q7. Comment faire pour accéder à l’état
désiré-? Liens entre ICH Q7, ICH Q9 et ICH Q10.
18. Les évolutions récentes en termes de textes et d’interprétation
19. Les futures évolutions et les textes en préparation
Un QCM vous est proposé afin d’évaluer les connaissances acquises.
Réglementation applicable aux principes actifs : API / ICH Q7
Programme
● Appréhender les réglementations inter-nationales, américaines, européennes et japonaises relatives aux bonnes pra-tiques de fabrication des principes actifs pharmaceutiques.
● Comprendre le rôle des différents dépar-tements industriels (production, contrôle qualité, R&D, assurance qualité) dans l’ap-plication des principes réglementaires.
● Comprendre les attentes des organismes officiels assurant les inspections et être capable de se préparer à ces inspections.
● Acquérir les connaissances réglementaires permettant d’assurer la qualité du produit.
● Savoir se mettre en conformité avec les exi-gences de l’ICH Q7 de façon pragmatique.
● Avoir une vue sur les possibilités d’évolu-tion des exigences dans le futur.
Objectifs
B32
FORMATION ACTUALISEE
Formation inter-entreprises
DUREE 2 jours
PUBLIC
Tout cadre des Industries de la Santé
fabricant et/ou utilisant des principes
actifs pharmaceutiques
B10
A15
B27
B104
B26
B8

QU
ALI
TE
/ R
EG
LEM
EN
TAT
ION
27Pour toute formation intra-entreprise : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
Les laboratoires de contrôle qualité se doivent d’avoir un système qualité ; mais quel système mettre en place et comment le faire vivre de façon pragmatique ? Comment mettre en œuvre les normes existantes et imposées ?
Module 1 : La Règlementation et les normes applicables à un laboratoire de contrôle qualitéLes autorités de tutelle : AFSSAPS, EMEA, FDALégislation pharmaceutique : enregistrement (AMM, NDA, DMF), inspection (BPF, BPL, BPC, BPLC).Législation des cosmétiques (BPPC), des dis-positifs médicaux, marquage CE. Normes ISO 9001, 17025, 22716. Les valeurs essentielles requises ou recommandées par tous pour la crédibilité et la fiabilité des données.
Module 2 : Assurance Qualité Système de management de la qualité.Rôles et Responsabilités de l’Assurance Qualité.Assurance Qualité (AQ) versus Contrôle Qualité (CQ) : quelles différences ?Le Manuel Qualité. Les composants princi-paux du programme AQ/QC.Les particularités pour les laboratoires parti-cipant aux essais cliniques.Personnel et gestion des formations (plan de formation, qualification).Maitrise des Procédés : - Indicateurs Qualité - Identification des dysfonctionnements - Cartes de contrôle, plans d’échantillonnage
Audits internes.Sous-traitance.
Module 3 : Contrôle QualitéQu’est ce que le contrôle qualité et où est-il implémenté dans le cycle de vie du produit?Les Activités du laboratoire (flux). L’organi-sation du laboratoire. Le cycle d’assurance qualité au laboratoire. L’indépendance par rapport à la production. Les Responsabilités de l’unité de Contrôle Qualité. Les principes de base du Contrôle Qualité
Module 4 : Les Documents CQMaîtrise des documents et bonnes pratiques documentaires : approbation, distribution, changement. Les procédures requises par les autorités. Les documents du CQ. Les Spé-cifications des produits : matière première, l’eau de procédé, produits finis, contrôle en cours. Maîtrise des enregistrements relatifs
à la qualité. La feuille de travail. Le cahier de laboratoire. Certificats et Rapports. Archivage et durée de rétention.
Module 5 : Locaux et Equipements (Analy-tique, Non Analytique) Spécifications des locaux : considérations spécifiques pour le CQ pour les essais physico-chimiques, microbiologiques, virologie, de stérilité, et la métrologie, Monitoring d’Envi-ronnement pour le CQ. Hygiène et Sécurité : risques chimiques, risques biologiques, Qua-lité de l’eau pour le laboratoire, Spécification des équipements et identification. Calibration ou Qualification ? Qualification versus valida-tion ?Validation des équipements automatisés et des systèmes informatisés : - Cycle de vie de la Qualification d’un équi-pement (QF, QD, QI, QO, QP),
- Cycle de vie de la validation système infor-matisé (QF, QD, QI, QO, QP),
- Exigences règlementaires concernant les enregistrements électroniques,
- La maitrise des changements,Nettoyage et maintenance.Conditions de stockage et enregistrements.
Module 6 : Bonnes Pratiques d’Echan-tillonnage et d’analyseAchats. Comprendre les erreurs lors des échantillonnages et analyses : risque produit, risque patient. Manipulation des échantillons soumis à l’essai. Technique d’Echantillo-nnage. Plan d’Echantillonnage. Identification et Traçabilité des échantillons. Qu’est qu’un échantillon témoin ? Réactifs/Références/Standards primaires : certificats, préparation, identification, étiquetage, péremption et traçabilité. Etude de stabilité : échantillons, plan de stabilité et rapport. Calibration, Véri-fication, Valeurs hors spécification, Certificats et Enregistrements. Validation des Méthodes Analytiques (CIH, Pharmacopées) : - Méthodes pharmacopées : à valider ou à vérifier ?
- Méthodes spécifiques, - Nettoyage verrerie, - Transfert des méthodes,
Traçabilité des tests,Vérification des résultats de tests. Maîtrise des non-conformités, des résultats hors spé-cifications OOS et processus d’investigation. Contrôle spécifiques : retours, retravaille, re-traitement, contrôle périodique.
EXERCICES DE MISE EN SITUATIONPlusieurs exercices sont proposés pendant la formation pour illustrer les points abordés.
Un QCM vous est proposé afin d’évaluer les
connaissances acquises.
Assurance Qualité et Contrôle Qualité dans les laboratoires de contrôle
Programme
● Comprendre les exigences GxP organi-sationnelles et opérationnelles (GCP, GLP, GMP, GCLP) applicables au laboratoire de Contrôle Qualité.
● Savoir comment détecter les pratiques impropres au laboratoire.
● Savoir mettre en place le plan d’AQ et de CQ.
Objectifs
B33FORMATION ACTUALISEE
Formation inter-entreprises
DUREE 2 jours
PUBLIC
Cette formation s’adresse aux Managers,
Superviseurs et au personnel qui seront
amenés à construire et maintenir un
système qualité pour les laboratoires de
contrôle.
B10
A15
Toutes nos formations sont
réalisables en intra-entreprise
Contactez-nous + 212 (0)6 61 13 69 28
www.2ssante.com

28
QU
ALI
TE
/ R
EG
LEM
EN
TAT
ION
Pour tout projet de formation : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
1. Le contexte réglementaireConnaître les exigences réglementaires
applicables à la revue qualité produit : BPF
(AFSSAPS), cGMP (FDA), GMP (EMEA).
Définition des termes employés dans la mise en
œuvre du processus de revue qualité produit.
2. Maîtrise du processus de revue qualité produit
2.1. La procédure de revue qualité produitRappel des principes généraux. Définir les
acteurs et leurs responsabilités. Principes de
collecte et traitement des données. Contenu
de la revue.
2.2. Format et règles de représentation du dossierLes différentes règles selon les formes galé-
niques.
Recueil des données :
- Production
- Contrôle
- Hors spécifications
- Hors tendances
- Ecarts/déviations critiques
- Réclamations et retours
- Revue des changements
- Etc.
Vérification de la conformité aux dossiers
d’enregistrement.
Mise en place des actions correctives / pré-
ventives.
2.3. Validation de la revue qualité produit et CAPA Maîtrise des procédés.
Modification/mise à jour des paramètres
critiques.
Validation des procédés.
Analyse d’impact sur écarts et déviations.
3. ConclusionPoints clés de la revue qualité produit :
- Revoir et maintenir la qualité des produits à
travers une surveillance active des données
qualités critiques.
- Evaluer si des actions correctives/préven-
tives peuvent être mise en place afin de cor-
riger ou d’améliorer la qualité des produits
et la conformité réglementaire (Processus
d’Amélioration Continu : PAC).
Un QCM vous est proposé afin d’évaluer les
connaissances acquises.
La revue qualité produit souvent appelée « APR » pour Annual Product Review » est devenue un outil de gestion de la qualité indispensable et de surcroît obligatoire du point de vue des instances réglementaires nationales et internationales. La sur-veillance active des données qualité critiques doit garantir le maintient de la qualité des produits et déclencher si nécessaire des programmes d’actions correctives ou d’actions préventives.
APR – Annual Product Review Revue Qualité Produit
Programme
● Connaître les exigences des réglementa-
tions BPF de l’AFSSAPS, cGMP de la FDA
et GMP de l’Europe concernant la revue
produit en tant qu’outil de gestion de la
qualité.
● Savoir maîtriser le processus de revue
qualité produit.
● Savoir identifier les éléments nécessaires
à la constitution du dossier.
● Savoir rédiger la procédure générale de
revue qualité produit.
● Savoir définir les actions préventives/
correctives (CAPA) à mettre en place.
Objectifs
B36NOUVEAU
Formation inter-entreprises
DUREE 1 jour
PUBLIC
- Assurance Qualité
- Contrôle Qualité
- Production
- Qualification / Validation
Vous souhaitez obtenir
plus d’informations ?
Contactez-nous + 212 (0)6 61 13 69 28
www.2ssante.com

QU
ALI
TE
/ R
EG
LEM
EN
TAT
ION
29Pour toute formation intra-entreprise : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
Module 1 : Rappel sur la Règlementation et les normes applicables à un laboratoire de contrôleLes autorités de tutelle : AFSSAPS, EMEA, FDA.
Législation pharmaceutique : BPF, BPL, BPLC
Normes ISO 9001, 17025, 22716.
Module 2 : Contrôle QualitéQu’est ce que le contrôle qualité et où est-il
implémenté dans le cycle de vie du produit?
Les activités du laboratoire (flux).
L’organisation du laboratoire.
Le cycle d’assurance qualité au laboratoire.
L’indépendance par rapport à la production.
Les responsabilités de l’unité de Contrôle
Qualité.
Les principes de base du Contrôle Qualité.
Module 3 : Les Documents CQMaîtrise des documents et bonnes pratiques
documentaires : approbation, distribution,
changement.
Les procédures requises par les autorités.
Les documents du CQ.
Les spécifications des produits : matière
première, l’eau de procédé, produits finis,
contrôle en cours. Maîtrise des enregistre-
ments relatifs à la qualité.
La feuille de travail.
Le cahier de laboratoire.
Certificats et rapports.
Archivage et durée de détention.
Module 4 : Equipements de laboratoire
Spécification des équipements et identifica-
tion.
Calibration ou Qualification ? Qualification
versus validation ?
Validation des équipements automatisés et
des systèmes informatisés :
- Cycle de vie de la Qualification d’un équipe-
ment (QF, QD, QI, QO, QP)
- Cycle de vie de la validation système infor-
matisé (QF, QD, QI, QO, QP),
- Exigences règlementaires concernant les
enregistrements électroniques,
- La maîtrise des changements.
Nettoyage et maintenance.
Conditions de stockage et enregistrements.
Module 5 : Bonnes Pratiques d’analyse
Identification et Traçabilité des échantillons
Qu’est qu’un échantillon témoin ?
Réactifs / Références / Standards primaires :
certificats, préparation, identification, étique-
tage, péremption et traçabilité.
Etude de stabilité : échantillons, plan de sta-
bilité et rapport.
Calibration, Vérification, Valeurs hors spécifi-
cation, Certificats et Enregistrements.
Validation des Méthodes Analytiques (CIH,
Pharmacopées) :
- Méthodes pharmacopées : à valider ou à
vérifier ?
- Méthodes spécifiques,
- Nettoyage verrerie,
- Transfert des méthodes.
Traçabilité des tests.
Vérification des résultats de tests.
Maîtrise des non-conformités, des résultats
hors spécifications OOS et processus d’inves-
tigation.
Contrôles spécifiques : retours, reprise,
contrôle périodique.
EXERCICES DE MISE EN SITUATION
Plusieurs exercices sont proposés pendant la
formation pour illustrer les points abordés.
Un QCM vous est proposé afin d’évaluer les
connaissances acquises.
Les laboratoires de chimie se doivent, comme les autres laboratoires dans le domaine de la santé, d’avoir un système qualité et de travailler selon de bonnes pratiques. Mais quelles sont les règles à appliquer et comment être certain de les respecter au quotidien ?
Les bonnes pratiquesdans les laboratoires de chimie
Programme
● Comprendre les exigences GxP organisa-
tionnelles et opérationnelles (GLP, GMP)
applicables au laboratoire de Contrôle
Qualité.
● Savoir appliquer et respecter ces règles
dans la vie quotidienne du laboratoire.
● Connaître et savoir respecter les bonnes
pratiques d’analyse.
Objectifs
B39
PUBLIC
Techniciens chimistes
B211
Pour développer vos compétences, nous vous conseillons également :
B32
NOUVEAU
A40
A131
Toutes nos formations sont
disponibles en anglais
Contactez-nous + 212 (0)6 61 13 69 28
www.2ssante.com

GESTION DES RISQUES

GE
ST
ION
DE
S R
ISQ
UE
S
31Pour toute formation intra-entreprise : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
1. Analyses de risques : définitions et théorie
Définition de la terminologie utilisée en
analyse de risques (causes, conséquences,
risques, défauts, erreurs, défaillances, …).
Description des différents types d’analyses
de risques (inductive, déductive … et AMDEC,
HAZOP, arbre de défaillance, …).
2. Pourquoi une méthode spécifique à la validation ?Rappel sur les attentes de la validation et le
but de l’analyse de risques.
3. Méthode CVO-RM : concept et théoriePrésentation du concept d’une méthodologie
analyse de risques applicable à la validation et
ses avantages.
Présentation d’une échelle unique de risques,
du choix du nombre de niveaux avec leurs
avantages et inconvénients.
4. La méthodologie CVO-RM : Présentation des 9 étapes de la méthodologie1. Sélectionner les références réglementaires
2. Sélectionner les exigences réglementaires
3. Déterminer les risques, les conséquences (et
leur criticité) et les causes génériques
4. Réaliser l’analyse de risques Processus
5. Réaliser l’analyse de risques Fonctionnelle
6. Réaliser l’analyse de risques Composant
7. Déterminer les éléments à qualifier
8. Déterminer les profondeurs de test
9. Générer les matrices de traçabilité
EXERCICES DE MISE EN SITUATION - Choix d’une échelle unique
- Choix de textes réglementaires
- Définition des risques et création d’un cata-
logue de risques
- Réalisation d’analyses de risques sur les
Processus, les Fonctions et les Composants
- Génération de cas de tests basés sur le
résultat des analyses de risques
- Génération de Matrice de Traçabilité
Un QCM vous est proposé afin d’évaluer les
connaissances acquises.
Les autorités réglementaires, et particulièrement la FDA, insistent sur l’importance d’une analyse de risques pour adapter les stratégies de validation et focaliser l’effort de validation sur les systèmes ou portions de systèmes qui le nécessitent. L’approche d’analyse de risques qui vous est proposée est un nouveau concept CyberConseil, simple, pragmatique et efficace : les risques sont d’abord déterminés par rapport aux exigences réglementaires et enregistrés dans un catalogue de risques puis on y ajoute les risques techniques liés au système lui-même. Ils sont ensuite utilisés pour sélectionner les processus, fonctions et composants à valider et définir la profondeur des tests à mettre en œuvre.
Analyse de Risques : CVO-RM (Risk Management)
Objectifs
● Connaître la terminologie utilisée en
analyse de risques, les standards et les
normes.
● Connaître les différentes méthodes
d’analyse de risques leurs avantages et
leurs inconvénients.
● Appréhender les différentes étapes de la
méthodologie : CVO-RM.
● Savoir définir une échelle unique de
risques.
Programme
B9
PUBLIC
Toute personne souhaitant apprendre
la méthode pragmatique d’analyse de
Risques CVO-RM de CVO-Cyberconseil
conçue pour les projets de validation et
de qualification.
B29
Pour développer vos compétences, nous vous conseillons également :
B30
Toutes nos formations sont
réalisables en intra-entreprise
Contactez-nous + 212 (0)6 61 13 69 28
www.2ssante.com

32
GE
ST
ION
DE
S R
ISQ
UE
S
Pour tout projet de formation : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
1. Enjeux, standards et réglementationPanorama des textes majeurs sur la gestion
des risques liée aux dispositifs médicaux.
Les démarches FDA et européennes en
matière de gestion des risques.
Quelles sont les obligations réglementaires
en termes de gestion des risques ?
Pourquoi la gestion des risques est-elle
nécessaire en dispositifs médicaux ?
2. DéfinitionsRappel des terminologies propres à la gestion
des risques. Différenciation entre analyse
« déductive » et « inductive », « qualitative » et
« quantitative », « processus » et « fonction-
nelle » ainsi qu’entre « danger » et « risque »,
« cause » et « erreur », …
3. ISO 14971:2007Présentation de l’ISO 14971:2007 « Applica-
tion de la gestion des risques aux Dispositifs
Médicaux » : contenu et principales exigences.
Revue détaillée des principaux chapitres de
l’ISO 14971:2007.
Différences entre la version 2007 et la version
2000.
4. Théorie de gestion des risquesLes concepts derrière la gestion des risques.
La terminologie utilisée en gestion de risques.
Les pré-requis à toutes analyses de risques.
Comment conduire une analyse de risques.
Savoir déterminer les points-clés pour réussir
une analyse de risques : définition de l’équipe,
choix des référentiels et du périmètre, déter-
mination des échelles d’évaluation.
Le contenu d’une analyse de risques.
Comment se déroule une analyse de risques ?
5. Processus de gestion des risquesAnalyse du Risque. Evaluation du Risque.
Maîtrise du Risque. Evaluation de l’acceptabi-
lité du risque résiduel global. Rapport de ges-
tion des risques. Information de production
et de postproduction.
6. Outils méthodologiquesPrésentation des différents types d’analyses
de risques (déductif, inductif, …).
Présentation détaillée des principaux outils
méthodologiques, de leurs avantages et de
leurs inconvénients, de leur application :
- Diagramme Cause-Effet
- AMDE / AMDEC
- HAZOP
- Arbre de Pannes
- CVO-RM
- HACCP
Présentation de la démarche analyse de
risques Produit.
Présentation de la démarche analyse de
risques Processus.
Dans la réglementation européenne et américaine relative aux dispositifs médicaux, la gestion des risques est définie comme un élément incontournable du dossier produit et des processus associés. Cependant, dans ces textes, ne sont décrits que les grands principes de la méthodologie à appliquer. Peu d’informations pratiques sont données sur les outils et les techniques utilisables mais l’ISO 14971:2007 est citée comme la référence et le guide à suivre pour la gestion des risques. Cette formation permet de faire un panorama des exigences réglementaires, une revue de l’ISO 14971:2007 et des méthodes d’analyses de risques disponibles. Puis d’appliquer les principes définis dans une gestion des risques adaptée aux dispositifs conçus et fabriqués dans votre entreprise.
Exercices pratiques et études de casQuel(s) type(s) d’analyse(s) de risques utiliser
dans votre entreprise ?
Détermination des critères d’analyse (Gravité,
Occurrence, Détection, Autres).
Détermination des méthodes d’évaluation et
de maîtrise des risques.
Cas pratique d‘analyse de risques sur
exemple(s) concret(s).
Un QCM vous est proposé afin d’évaluer les
connaissances acquises.
La gestion des risques appliquée aux Dispositifs Médicaux (ISO 14971:2007)
Objectifs
● Connaître les exigences réglementaires
et normatives de la CE et de la FDA.
● Savoir pourquoi les analyses de risques
sont nécessaires.
● Connaître les exigences de l’ISO
14971:2007 et les différences avec la
version 2000.
● Connaître la terminologie utilisée en
gestion des risques.
● Connaître les concepts liés à la gestion
des risques.
● Connaître les principales méthodologies
d’analyse de risques.
● Savoir quel type d’analyse de risques
utiliser en fonction du but recherché.
● Acquérir une méthodologie pragmatique
de l’analyse de risques.
● Savoir définir des méthodes de gestion
des risques.
Programme
B29
PUBLIC
Toute personne impliquée dans la gestion
des risques concernant les Dispositifs
Médicaux.
B28
Pour développer vos compétences, nous vous conseillons également :
B9Vous souhaitez obtenir
plus d’informations ?
Contactez-nous + 212 (0)6 61 13 69 28
www.2ssante.com

GE
ST
ION
DE
S R
ISQ
UE
S
33Pour toute formation intra-entreprise : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
Dans la réglementation européenne et américaine relative au domaine de la Santé, la gestion des risques est définie comme un élément incontournable du dossier produit et des processus associés. Cependant, dans ces textes, ne sont décrits que les grands principes de la méthodologie à appliquer. Peu d’informations pratiques sont données sur les outils et les techniques utilisables mais l’ICH Q9 est désormais la référence et le guide à suivre pour la gestion des risques. Cette formation permet de faire un panorama des exigences réglementaires, une revue de l’ICH Q9 et des méthodes d’analyses de risques disponibles. Puis d’appliquer les principes définis dans une gestion des risques adaptée aux produits et aux processus de votre entreprise.
1. Enjeux, standards et réglementationPanorama des textes majeurs sur la gestion
des risques dans le domaine de la santé.
Les démarches FDA et européennes en ma-
tière de gestion des risques.
Quelles sont les obligations réglementaires
en termes de gestion des risques ?
Pourquoi la gestion des risques est-elle né-
cessaire dans le domaine de la santé ?
2. DéfinitionsRappel des terminologies propres à la
gestion des risques. Différenciation entre
analyse « déductive » et « inductive », « qualita-
tive » et « quantitative », « processus » et « fonc-
tionnelle » ainsi qu’entre « danger » et « risque »,
« cause » et « erreur », …
3. ICH Q9Présentation de l’ICH Q9 « Gestion des risques
qualité » : contenu et principales exigences.
Revue détaillée des principaux chapitres de
l’ICH Q9.
4. Théorie de gestion des risquesLes concepts derrière la gestion des risques.
La terminologie utilisée en gestion de risques.
Les pré-requis à toutes analyses de risques.
Comment conduire une analyse de risques.
Savoir déterminer les points-clés pour réussir
une analyse de risques : définition de l’équipe,
choix des référentiels et du périmètre, déter-
mination des échelles d’évaluation.
Le contenu d’une analyse de risques.
Comment se déroule une analyse de risques ?
5. Processus de gestion des risques selon l’ICH Q9Estimation du risque : Identification du risque,
Analyse du risque, Evaluation du risque.
Maîtrise du risque : Réduction du risque,
Acceptation du risque.
Revue du risque : Revue des évènements.
6. Outils méthodologiquesPrésentation des différents types d’analyses
de risques (déductif, inductif…).
Présentation détaillée des principaux outils
méthodologiques, de leurs avantages et de
leurs inconvénients, de leur application :
- Diagramme Cause-Effet
- AMDE / AMDEC
- HAZOP
- Arbre de Pannes
- CVO-RM
- HACCP
Présentation de la démarche analyse de
risques Produit. Présentation de la démarche
analyse de risques Processus.
Exercices pratiques et études de casQuel(s) type(s) d’analyse(s) de risques utiliser
dans votre entreprise ?
Détermination des critères d’analyse (Gravité,
Occurrence, Détection, Autres).
Détermination des méthodes d’évaluation et
de maîtrise des risques.
Cas pratique d‘analyse de risques à partir
d’exemple(s) concret(s).
Un QCM vous est proposé afin d’évaluer les
connaissances acquises.
La gestion des risques dans le domaine de la Santé (ICH Q9)
Objectifs
● Connaître les exigences réglementaires
et normatives de la CE et de la FDA.
● Savoir pourquoi les analyses de risques
sont nécessaires.
● Connaître les exigences de l’ICH Q9.
● Connaître la terminologie utilisée en
gestion des risques.
● Connaître les concepts liés à la gestion
des risques.
● Connaître les principales méthodologies
d’analyse de risques.
● Savoir quel type d’analyse de risques
utiliser en fonction du but recherché.
● Acquérir une méthodologie pragma-
tique de l’analyse de risques.
● Savoir définir des méthodes de gestion
des risques.
Programme
B30
Formation inter-entreprises
DUREE 2 jours
PUBLIC
Tout cadre des industries de la Santé,
quelque soit son domaine d’activité (sauf
les industriels des Dispositifs Médicaux
pour qui la formation B29 est mieux
adaptée).
B9
Vous souhaitez réaliser une
formation sur l’ICH Q8 ?
Contactez-nous + 212 (0)6 61 13 69 28
www.2ssante.com

34
GE
ST
ION
DE
S R
ISQ
UE
S
Pour tout projet de formation : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
Les OOS sont au cœur d’un important enjeu business : faut-il retraiter les lots, les jeter, les déclasser ? Quelle que soit la nature des actions correctives et/ou des actions préventives entreprises, les OOS sont potentiellement signe d’un manque de maitrise du processus. C’est la raison pour laquelle, au-delà de l’enjeu business, un enjeu réglementaire tout aussi important est positionné. D’autant plus que ces OOS et leurs CAPAs associées sont un point d’entrée très apprécié des inspecteurs.Comment les identifier, les traiter, enregistrer les actions prises, vérifier l’efficacité des actions, mettre en place puis suivre les indicateurs, … sont autant de points abordés dans cette formation.
1. RéglementationRappel sur la réglementation liée aux OOS et
aux CAPA.
2. OOS : C’est quoi ?Définitions.
Terminologie autour des OOS.
3. CAPA : C’est quoi ?Définitions.
Terminologie autour des CAPA.
4. Relations OOS / CAPA / Validation / IPCRelations OOS / CAPA.
In Process Control.
Relations Validation / IPC / OOS.
5. Processus Gestion OOS / CAPAProcessus de gestion des OOS / CAPA.
Importance des investigations.
Indicateurs qualité et tableaux de bord.
6. Analyse d’Impacts / Analyse des CausesProcessus d’analyse d’impacts.
Processus d’analyse des causes.
7. OOS / CAPA … La réalité financière et les intérêts de l’entreprise Satisfaction client et succès de l’entreprise.
Les risques à maîtriser.
8. Les révolutions culturelles qualité… Douleurs, mais bénéfices
Le coût de la non-conformité.
Contrôle des modifications versus gestion
des modifications.
9. L’ultime but … La satisfaction des clientsCAPA au centre du problème …
Analyse de risques au centre de la solution …
EXERCICES DE MISE EN SITUATIONApplication Pratique OOS / CAPA.
Un QCM vous est proposé afin d’évaluer les
connaissances acquises.
OOS/CAPA – Maîtriser vos Hors Spécificationset optimiser vos Actions Correctives
Objectifs
● Connaître le processus de gestion des
OOS et des CAPA.
● Savoir reconnaître et traiter les OOS.
● Savoir faire la différence entre Correction,
Action Corrective et Action Préventive.
● Connaître les interactions entre Valida-
tion, OOS et IPC.
● Connaître les risques et les enjeux liés à
la gestion et la non gestion des OOS et
CAPA.
● Connaître les points clés d’une structure
qui permette de garantir la satisfaction
client et réglementaire tout en restant
pragmatique.
Programme
C13
PUBLIC
- Membres de l’Assurance Qualité
- Membres du Contrôle Qualité
- Chefs de Produit (au sens technique)
- Responsables de lignes de production
Des formateurs experts et terrain
Des formations pragmatiques
Une veille réglementaire quotidienne
En phase avec l’évolution de vos métiers
Totale indépendance
Dédié à un seul métier : la Santé
Support de formation détaillé et complet
Nombre de stagiaires limité
Contrôle des connaissances
Qualité de l’accueil
10 RAISONS DE CHOISIR LES FORMATIONS CVO-CYBERCONSEIL

GE
ST
ION
DE
S R
ISQ
UE
S
35Pour toute formation intra-entreprise : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
1. IntroductionPrésentation de la journée de formation. Un point important va être accordé sur la séman-tique des mots utilisés dans le monde de la gestion des risques pour lever toute ambiguïté par la suite.
2. Les étapes de la gestion des risques.Nous allons balayer dans ce chapitre les grandes étapes incontournables pour réaliser avec succès votre programme de gestion de risques. Les étapes de la gestion de risques :2.1.Le diagnostic des risques : Apprendre à identifier les vulnérabilités (humaines, techniques, informationnelles, financières et liées aux partenaires) et les menaces (humaines, naturelles, techno-logiques, politiques, pays, sociétales, etc.), à réfléchir sur les objectifs de l’entreprise avant et après sinistre, déterminer le niveau acceptable de risque et mesurer l’impact financier.2.2.Etudier les outils pour traiter les risques : Comment réduire leur impact et leur fréquence, comment financer les pertes, ou comment transférer le risque à un tiers comme l’assureur.2.3.L’élaboration du programme de ges-tion de risques : Le montage du programme passera par l’évaluation financière du projet, le calcul du retour sur investissement, et cela en fonction des objectifs de l’entreprise et de sa stratégie.2.4.La mise en œuvre : Elle demandera une étude plus technique et approfondie pour choisir les moyens et équi-pement à mettre en place. Nous balayerons les différentes technologies existantes en fonction des problématiques.
3. L’établissement des prévisions.Rappel sur les calculs de statistiques et de tendances pour permettre ensuite l’évaluation de la sinistralité.
4. Calculs financiers.En complément des calculs statistiques, ceux-ci permettront d’estimer le retour sur investissement afin de le comparer à celui exigé par les services financiers.
5. Définir les objectifs de son programme de gestion de risques.Pour réussir son projet de gestion de risques, cela passe par la définition des objectifs de l’entreprise avant et après sinistre. Nous prendrons également en compte le niveau d’acceptation du risque par l’équipe dirigeante en rapport avec la stratégie du groupe. Ceci définira le substrat du programme de gestion de risques.
6. Les outils pour réduire l’incertitude. Lors de cette étape, nous allons voir l’ensemble des concepts permettant de réduire la gravité des sinistres et la fréquence des aléas. Des exemples viendront enrichir la présenta-tion.
7. Les moyens pour financer les consé-quences des aléas et sinistresNous verrons que dans certaines situations, il n’existe pas de solution pour réduire l’impact d’un sinistre, il est alors plus efficient de le prendre en charge par la trésorerie courante de l’entreprise, plutôt que d’investir inuti-lement dans des moyens excessivement chers. Dans d’autres cas l’évènement étant peu probable et la gravité tellement élevée,
Concept né il y a plus de 60 ans aux États-Unis, le Risk Management arrive aujourd’hui en France avec une grande maturité. La crise économique a mis en exergue l’intérêt de cet outil pour réduire l’incertitude et l’impact financier lors d’aléas qui touchent les organisations. Au-delà de la réduction des risques, le Risk Management permettra de lisser les résultats financiers même en cas de sinistres graves. Dans les mois à venir, les agences de notations vont être amenées à s’intéresser de plus en plus à la manière dont les entreprises gèrent leurs risques. L’affaire des Subprimes où les conséquences ont largement débordé sur l’économie mondiale a été le principal élément déclencheur de cette démarche.
l’assurance sera alors le moyen le plus sûr pour transférer le risque.
8. La gestion de crise et les plans de conti-nuitéParfois, les risques sont non assurables et la protection est également impossible à mettre en œuvre : le seul outil pour réduire l’impact est alors la mise en place d’un plan de crise et d’un plan de continuité.
9. Le suivi du programme de gestion de risques.Le manager devra animer son pôle de Risk management à l’aide de différents tableaux de bord, et réactualiser la politique globale en fonction des nouveaux objectifs.
EXERCICES DE MISE EN SITUATIONPlusieurs exercices permettront de revoir l’ensemble de la méthode et des calculs de statistiques et financiers.
Un QCM vous est proposé afin d’évaluer les connaissances acquises
Mettre en place une méthode de Risk Management dans votre laboratoire pharmaceutique
Objectifs
● Apprendre une méthode.
● Identifier les vulnérabilités de l’organisa-tion.
● Identifier les menaces (risques juridiques, naturels, industriels, humains, etc.).
● Mettre en place des outils d’enregistre-ment de la sinistralité pour évaluer le poids annuel financier du risque.
● Déterminer les objectifs de l’entreprise avant sinistres et après sinistres.
● Trouver le bon compromis entre les projets de prévention, de protection ou d’assurance.
● Evaluer le retour sur investissement de vos projets.
● Découvrir la gestion de crise : Plan de crise, cellule de crise et communication de crise. Construire un plan de continuité.
● Suivre la sinistralité de votre organisation.
● Renégocier vos contrats d’assurance en couvrant tout le spectre de vos risques.
Programme
C58
NOUVEAU
PUBLIC
- Responsables Qualité
- Responsables de Services Généraux-
Chefs de projet sur un risque particulier
- Responsables des Systèmes
d’Information
- Personnels HSE
- Risk Managers débutants.
Le module peut être adapté sur un
thème de risques particuliers en fonction
du groupe.
PRE-REQUIS
Avoir des bases en comptabilité, finance
et en droit de la responsabilité civile. Le
niveau en mathématique nécessaire lors
de cette formation est celui de terminal.
La démarche pédagogique est calquée
sur le mode anglo-saxon, avec de
nombreux aller-retour aux travers des
concepts pour ainsi vous permettre
d’assimiler la méthode.
C59

36
GE
ST
ION
DE
S R
ISQ
UE
S
Pour tout projet de formation : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
1. Points clés et enjeux - Définitions et objectifs de la gestion des risques informatiques et métiers, des réponses apportées par le BCP et le DRP.
- Typologie des sinistres. - L’évolution des entreprises et de leur stra-tégie : l’importance de la disponibilité de l’information et des systèmes.
1.2. Les réglementations, les guides et les normes - Description des exigences et propositions des référentiels français, européens, nord-américains et internationaux : GMP, GLP, GCP, 21 CFR Part 11, BPF, BPL, BPC, BPD, ICH, GAMP, PIC/S, normes ISO-NF, BS, ITIL.
- Position des agences officielles. Points-clés d’une inspection : AFSSAPS, FDA, EMEA.
- Analyse des textes à l’aide de Warning Letters
et/ou de 483’s de la FDA. - Contraintes réglementaires et exigences : recommandations ISO, normes Bâle II, contrats d’assurances.
1.3 Les principes de base - Introduction à la notion de disponibilité des systèmes d’information dans les industries de la santé.
- Concepts : la sensibilité des activités à la non disponibilité des données et définition des points critiques.
1.4 BCP – DRP et Maturité - Présentation du SMM (Security Maturity Model).
- Comment déterminer le niveau de maturité de votre entreprise ?
- Impacts de la maturité sur les projets infor-matiques.
Les entreprises subissent une dépendance croissante vis-à-vis de la technologie en raison de l’élargissement du champ d’applica-tion des nouvelles technologies de l’information et de la communication dans leur organisation, ainsi que de l’évolution continue des systèmes d’information. L’accroissement, l’instantanéité et la globalisation des échanges d’informations rendent les activités opérationnelles des entreprises potentiellement vulnérables même dans le contexte d’un incident mineur touchant la disponibilité, l’accès aux données ou aux sites. Dans un monde de plus en plus sécurisé ou l’aléa n’est plus toléré, les dirigeants, les actionnaires, les instances réglementaires et les assureurs sont aujourd’hui de plus en plus préoccupés par la qualité des réponses que la gouver-nance d’entreprise apporte à cette problématique. L’objectif de cette formation est de proposer une réponse concrète, opérationnelle et modulable à ces attentes. Cette formation présente le management des risques relatifs à la disponibilité de l’information, des systèmes et des activités métiers, et propose des méthodologies pour maîtriser ces risques au travers de la mise en œuvre du plan de secours et de reprise de l’activité en cas de désastre (DRP - Disaster Recovery Plan), et du Plan de Continuité des Activités (PCA) de l’Entreprise (BCP - Business Continuity Plan).
Gestion des risques informatiquesBCP / DRP
Objectifs
● Connaître les exigences réglementaires applicables aux BCP et DRP.
● Comprendre l’environnement réglemen-taire pour être en conformité avec les exigences de l’AFSSAPS, de l’EMEA, de la FDA (BPF/GMP, BPL/GLP, BPC/GCP, BPD/GDP, ICH Q9…), de l’administration amé-ricaine (lois Sarbanes Oxley - SARBOX), des administrations européennes (lois sur les finances).
● Connaître les exigences normatives ap-plicables aux BCP et DRP : apport des référentiels ISO 23399, ISO 24762, ISO 27001 (ex 17799), BS 25999 (Business Continuity Institute), DRII, ITIL V3, GAMP 5, COBIT, ...
● Savoir définir une politique DRP adaptée à un environnement informatisé et auto-matisé.
● Savoir définir une politique BCP adaptée aux activités opérationnelles et aux métiers.
● Organiser le management des risques in-formatiques à l’échelle d’un ou plusieurs sites, d’un pays, d’un groupe et définir les exigences pour un BCP et un DRP.
● Sélectionner les architectures informa-tiques et solutions techniques les plus pertinentes.
● Mettre en place un DRP répondant aux risques techniques : spécifier, mettre en œuvre, vérifier, ajuster, maintenir.
● Formaliser un BCP répondant aux risques métiers : spécifier, mettre en œuvre, vérifier, ajuster, maintenir.
● Savoir définir et optimiser l’effort de mise en œuvre d’un BCP et/ou d’un DRP en fonction du contexte métier, et répartir les tâches entre les différents acteurs (valida-tion, qualité, informatique, ingénierie, …).
● Savoir auditer un BCP et/ou un DRP en cas d’externalisation d’activité informa-tique ou métier.
Programme
C59NOUVEAU
Formation inter-entreprises
DUREE 2 jours
PUBLIC
- Responsable Informatique
- Chef de Projet utilisateur
- Chef de Projet informatique
- Responsable Gestion des Risques - Risk
Manager
- Responsable sécurité
- Ingénieurs système & télécommunication
- Architectes des systèmes d’information
- Auditeurs internes ou externes
- Consultants
- Responsable de domaine
- Responsable Qualité
- Responsable Validation/Qualification
PÉRIMÈTRE
• Systèmes informatisés
• Systèmes automatisés
• Bureautique
• LAN, SAN, WAN
• Télécommunications
• Activités de Recherche & Développe-ment,
• Activités de laboratoire préclinique et clinique
• Activités de production
• Activités de distribution
• Activités commerciales
• Activités Réglementaires (vigilance, information,…)

GE
ST
ION
DE
S R
ISQ
UE
S
37Pour toute formation intra-entreprise : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
2. Le plan de secours et de reprise de l’acti-vité en cas de désastre : DRP2.1 Les parades au désastre technique : solutions organisationnelles, technolo-giques et physiques - Prendre en compte les besoins métiers : les scenarii à prendre en compte.
- L’analyse des risques techniques : système et réseaux.
- Les salles informatiques de backup, ap-proche multi-sites, les sites de reprise (hot, warm, cold sites, reciprocal agreement...)
- Les architectures de continuité de service : réseaux LAN, WAN .
- Les architectures de continuité de service : serveurs, clustering, load balancing, NAS.
- Les architectures de continuité de service : données, miroirs et techniques de réplica-tion de données (synchrone, asynchrone), SAN.
- La virtualisation. - Les solutions de sauvegarde et restauration - Les solutions d’archivage (bases de données, messagerie, bureautique, équipements de process, données métiers et entreprise...).
- Les technologies de reprise après sinistre. - Les réponses des solutions aux objectifs de temps de reprise (RTO - Recovery Time Objective) et de point de reprise (RPO - Reco-very Point Objective).
2.2 Conduire un projet DRP – Cycle de vie du DRP - Organiser le projet DRP : étude d’opportu-nité, les acteurs, le plan qualité, les comités de suivi.
- Externalisation ou traitement en interne du DRP.
- Les phases du projet DRP. - Le DRP : objectifs de reprise, la portée du plan, les procédures, la matrice des systèmes, la traçabilité avec l’analyse de risque et la réglementation.
- La contractualisation interne (SLA/OLA) ou externe (UC), les procédures, les responsa-bilités.
- Le DRP au quotidien. - La vérification des RTO & RPO et les tests périodiques du DRP (et des partenaires en cas d’externalisation).
- Les résultats des tests, les matrices et la correction du DRP.
- Maintenir un DRP actif. - L’audit du DRP.
3 – Le Plan de Continuité des Activités PCA / BCP – Cycle du vie du BCP - Prendre en compte les besoins métiers : les scenarii à intégrer.
- Organiser un projet BCP : étude d’opportu-nité, les acteurs, le plan qualité, les comités de suivi.
- Les phases du projet BCP. - Le BCP : objectifs de reprise, portée du plan, les procédures, la matrice des métiers, la traçabilité avec les systèmes, les technolo-gies et l’analyse de risque.
- Le plan de continuité : Gantt. - L’organisation du plan de continuité : la cellule de crise, les acteurs, les équipes les responsabilités.
- Les procédures d’escalade et les niveaux d’escalade.
- Les procédures informatiques, métiers, communicatio, ...
- La formation des acteurs. - le plan de secours activé : exploitation en mode secours et retour à la normale.
- La vérification des RTO & RPO et les tests périodiques du BCP.
- Les résultats des tests, les matrices et la correction du BCP.
- La maintenance / la non régression du BCP. - L’audit du BCP.
EXERCICES DE MISE EN SITUATION
Un cas pratique sous forme d’un fil rouge est déroulé tout au long de cette formation autour du système d’information d’une en-treprise ; les phases cibles présentées com-prennent : - analyse de risques métier - analyse de risques infrastructure informa-tique
- DRP - BCP
Un QCM vous est proposé afin d’évaluer et valider les connaissances acquises.
Gestion des risques informatiques – BCP / DRPC59
NOUVEAU
C58
Pour développer vos compétences, nous vous conseillons également :
B9C61
C60
Toutes nos formations sont
réalisables en intra-entreprise
Contactez-nous + 212 (0)6 61 13 69 28
www.2ssante.com

38
GE
ST
ION
DE
S R
ISQ
UE
S
Pour tout projet de formation : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
1. Introduction.Présentation de la journée de formation. Définition du vocabulaire lié à la sûreté, la malveillance, à l’espionnage industriel, qui sera utilisé tout au long de ce module.
2. La malveillance en France en faits et en chiffres.Indispensable à la prise en compte des risques, cet état des lieux, étayé d’exemples « démar-qués » vous permettra de bien assimiler les enjeux de votre société face à un problème croissant qui malheureusement n’arrive pas qu’aux autres. A la différence d’un sinistre incendie, par exemple, où les conséquences sont immédiatement visibles, les effets de l’espionnage industriel sont généralement décelés à retardement, avec le déni de res-ponsabilité du management.
3. L’inventaire des types d’actes malveillants.L’objectif de ce chapitre est de dresser en-semble un inventaire, le plus exhaustif pos-sible, des actes malveillants dans les activités de R&D, production, partenariat d’entreprise, et les phases de commercialisation et com-munication. Nous traiterons aussi bien du vol simple, que de l’espionnage industriel, du sabotage, de l’incendie volontaire, du ré-engineering social fréquemment utilisé comme cheval de Troie en sécurité informa-tique, ou encore du « piégeage » des salles de réunions à l’aide de micros. Dernier point, nous aborderons la problématique grandis-sante de la guerre de l’information.
4. Les motivations de la malveillance.Pour être en mesure de dresser des mu-railles face aux actes malveillants, il est in-
dispensable de s’intéresser à l’origine des motivations qui poussent des individus ou des concurrents à passer à l’acte. Connaître la récurrence dans la méthodologie straté-gique agressive de vos futurs partenaires ou de vos concurrents, c’est anticiper les risques probables
5. Evaluer vos risques malveillance.Nous proposons ici une méthode simple pour faire une première évaluation de vos risques au sein de votre établissement. Nous débu-terons par apprendre à faire le tour périmé-trique du site pour détecter les faiblesses dans vos clôtures. Indispensable aussi : apprendre à penser comme une personne malveillante et non comme individu qui respecte les règles et usages. Etape par étape, vous apprendrez à évaluer les différentes faiblesses.
6. Les moyens et technologies pour réduire les risques.Nous aborderons le trinôme indissociable en sûreté : moyens humains, moyens tech-niques et procédures à mettre en place. De la protection physique du site, en passant par la vidéo surveillance et ses contraintes régle-mentaires, nous verrons ensemble la mise en place d’un système de contrôle d’accès et ses contraintes sociales, la sensibilisation aux ap-pels téléphoniques suspects, …
7. Protéger l’information sensible tout au long de son cycle de vie.De la naissance de l’information à sa fin de vie, il est indispensable de lui donner un ni-veau de sensibilité et donc de confidentialité. Comment doit-on classifier le niveau de cette information, comment la protéger, qui en a « le besoin d’en connaître », et comment habi-
A l’heure de la crise mondiale, où la concurrence est de plus en plus exacerbée, l’arrivée de nouveaux entrants sur le marché, est facteur d’aggravation des potentiels actes de malveillance et d’espionnage industriel. Des concurrents de plus en plus agressifs ont tout intérêt dans le cadre de leur réduction des coûts en R&D de bénéficier des résultats de vos études en pratiquant l’art du renseignement et de la déstabilisation, avec parfois l’aide de médias eux-mêmes manipulés. Les techniques de minage des brevets, du lobbying normatif, et également le problème de la contrefaçon seront abordés.
liter les personnes. Reconnaître où se trouve l’information sensible et identifier quel est son cheminement dans l’organisation pour ainsi mieux la protéger. A l’issue comment détruire durablement l’information sensible avant qu’elle ne tombe entre de mauvaises mains.
8. Evaluer le coût de votre mise en sûreté.La phase clé de la mise en sûreté de votre site consiste à l’évaluation financière de votre pro-jet. Dans ce chapitre nous vous proposerons des ratios de coûts par élément de votre futur dispositif de sûreté.
9. La mise en place d’indicateurs de suivi.Quel est l’intérêt des indicateurs de suivi dans votre politique à long terme de la sûreté du laboratoire ? Comment les indicateurs vont-ils permettre de motiver les investissements dans vos projets de sûreté ? Quels indicateurs choisir pour votre tableau de bord au quoti-dien ? Comment les indicateurs, vont-ils être révélateurs de signaux faibles sur des tenta-tives de fraude ou d’intrusion ?
EXERCICES DE MISE EN SITUATIONPlusieurs exercices permettront de revoir la typologie des actes malveillants et les moyens pour les réduire.
Un QCM vous est proposé afin d’évaluer les connaissances acquises
La protection contre la malveillance et l’espionnageindustriel des laboratoires pharmaceutiques
Objectifs
● Faire l’inventaire des risques malveillants probables.
● Apprendre à connaître les méthodes uti-lisées par les services de renseignements privés pour mieux s’en prémunir.
● Comment procèdent les antivivisection-nistes pour assaillir ou bloquer votre site.
● Identifier les signaux faibles de la guerre de l’information ayant pour but de désta-biliser votre laboratoire.
● Apprendre à connaître les technologies de lutte contre la malveillance.
● Apprendre à gérer l’information sensible.
● Trouver le bon compromis entre les moyens humains et technologiques.
● Dresser un plan de mise en sûreté de votre site.
● Evaluer les coûts du programme de mise en sûreté.
Programme
C60
NOUVEAU
PUBLIC
- Responsables Qualité
- Responsables de Services Généraux
- Chefs de projet sur un risque particulier
- Responsables des Systèmes
d’Information
- Personnels HSE
- Risk Managers débutants
Le module peut être adapté sur un
thème de risques particuliers en fonction
du groupe.

39Pour toute formation intra-entreprise : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
Un accompagnement adapté
Étape indispensable dans tous
les plans de formation, l’accom-
pagnement permet de mesurer
l’impact, l’assimilation et la qualité
de la démarche.
Notre équipe formation a mis
en place un dispositif adapté à
chaque domaine et facilite ainsi
l’atteinte des objectifs fixés.
Afin de répondre au plus près
aux besoins de nos clients, nous
avons adopté une méthodologie
de construction de nos formations
basée sur le modèle d’instruction
ISD_ADDIE :
niveau 1 Connaissance
● information ● initiation ● notions générales
Personnes concernées : - Décideurs - Managers - Personnes motivées
niveau 2 Compréhension
● savoir ● comprendre et réagir ● études de situation
Personnes concernées : - Responsables Validation - Chefs de projet - Encadrement
niveau 3 Capabilité
● savoir-faire ● pratiquer ● cas pratiques
Personnes concernées : - Techniciens - Opérationnels
Méthodologie de construction de nos formations
Que doit-on enseigner ?
Comment doit-on l’enseigner ?
Préparer les outils de formation
Déployer la formation
Vérifier l’efficacité de la formation
et maintenir
Analysis
Design
Development
Implementation
Evaluation & Maintenance
Un accompagnement pédagogique spécifique
Des approches pédagogiques spécifiques
Notre Centre de Formation a développé des
approches pédagogiques spécifiques afin de
répondre à vos attentes. Les formations sont
ainsi adaptées aux publics visés, de la formation
sensibilisation à la formation action intégrant
des cas concrets.
Des formations concrètes pour acquérirde véritables compétences
Les formations CVO-CyberConseil sont élaborées avec des
cas concrets pour vous aider à acquérir les compétences
dont vous avez besoin rapidement et de manière approfondie.
Nos formations sont conçues pour vous apporter un retour
sur investissement immédiat.

40
GE
ST
ION
DE
S R
ISQ
UE
S
Pour tout projet de formation : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
La sécurité des systèmes d’information est et reste une préoccupation. De nombreux sinistres ces dernières années ont ravivé l’inquiétude. La couverture de ces sinistres est extrêmement vaste. On y trouve en effet des perturbations climatiques (inondation, canicule, tempête, panne électrique, ...), des attaques de hackers, des virus et des vers, des tentatives de récupération de données bancaires, des attaques terroristes, ... Les éléments marquants de ces sinistres sont leur importance et la rapidité avec laquelle ils se sont développés et transmis. L’importance du système d’informations n’a pour autant pas diminué et la mise en place de plus en plus large des processus de dématérialisation va encore accentuer le phénomène. Dans ce contexte, la sécurité des systèmes d’information voit son importance s’accroître au sein des entreprises.
1. Les dimensions du management de la sécurité1.1 Détermination des buts à atteindre - Comment intégrer les différentes compo-santes …
o Managérialeo Economique et financièreo Techniqueo Juridique et réglementaireo Organisationnelleo Environnementaleo Humaine … dans la définition d’une politique cible.1.2. Les entrées et les enjeux du manage-ment des risques - Description des exigences et propositions des référentiels français, européens, nord-américains et internationaux : GMP, GLP, GCP, 21 CFR Part 11, BPF, BPL, BPC, BPD, ICH, GAMP, PIC/S, normes ISO-NF, BS, ITIL.
- Position des agences officielles. Points-clés d’une inspection. AFSSAPS, FDA, EMEA.
- Analyse des textes à l’aide de Warning Letters et/ou de 483’s de la FDA.
- Contraintes réglementaires & exigences-: recommandations ISO, normes Bâle II, contrats d’assurances.
- Quels sont les référentiels normatifs et réglementaires à prendre en compte ? AFSSAPS, EMEA, FDA, …
- Quels sont les standards du domaine à prendre en compte ? ISO27001, ISO27002, ITIL v3, …
2. Détermination de la stratégie de sécuri-sation des systèmes informatisés2.1 La gouvernance d’entreprise - Comment intégrer la gestion de la sécurité dans la politique globale de management d’une entreprise, d’une direction informa-tique ?
- Quels sont les aspects juridiques, les risques légaux : LSF, Sarbanes-Oxley, CNIL, Bâle II …
Gestion de la sécurité des systèmes d’information
Objectifs
● Savoir comment considérer la sécurité des systèmes d’information comme une problématique complexe et l’envisager de manière systématique et globale.
● Savoir déterminer une approche globale qui intègre les composantes : stratégique, managériale, organisationnelle, juridique, technique et humaine, en considérant les facteurs risques, coûts et efficacité.
● Organiser le management des risques informatiques.
● Savoir développer les trois axes que sont : - la gouvernance d’entreprise - la gestion de la sécurité - la méthodologie et les technologies de la sécurité
● Comprendre les enjeux de l’entreprise.
● Savoir identifier les aspects juridiques et les risques légaux.
● Apprendre à aligner les objectifs opéra-tionnels sur les orientations stratégiques de l’entreprise.
● Acquérir la maîtrise des outils de mana-gement et de gouvernance des systèmes d’informations dans le domaine de la gestion de la sécurité.
● Assimiler les référentiels normatifs et réglementaires pour être en conformité avec les exigences de l’AFSSAPS, de l’EMEA, de la FDA (BPF/GMP, BPL/GLP, BPC/GCP, BPD/GDP, ICHQ9…), de l’admi-nistration américaine (SARBOX lois Sar-banes Oxley).
● Savoir appliquer les standards du do-maine : ISO27001, ISO27002, ISO2700x, BS25999, ITIL v3, GAMP 5, GAMP IT, COBIT, …
● Savoir définir et mettre en œuvre des politiques et des guides de sécurité : EBIOS, MEHARI, …
● Savoir mettre en place des plans de sensi-bilisation et de formation à la sécurité des systèmes d’informations.
● Savoir définir et mettre en œuvre un système.
Programme
C61NOUVEAU
Formation inter-entreprises
DUREE 2 jours
PUBLIC
- Directeur Informatique
- Risk Manager
- Responsable sécurité
- Ingénieurs système & télécommuni-
cation
- Auditeurs internes ou externes,
- Consultants
- Responsable Qualité,
- Responsable Validation/Qualification
- Ainsi que des cadres d’autres domaines
ayant en charge tout ou partie d’une
fonction « sécurité »
PÉRIMÈTRE
• Systèmes informatisés
• Systèmes automatisés
• Bureautique
• LAN, SAN, WAN
• Télécommunications
• Activités de Recherche & Développe-ment,
• Activités de laboratoire préclinique et clinique
• Activités de production
• Activités de distribution
• Activités commerciales
• Activités Réglementaires (vigilance, information, …)

GE
ST
ION
DE
S R
ISQ
UE
S
41Pour toute formation intra-entreprise : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
2.2. La politique sécurité - Comment définir et déployer une politique de sécurité informatique ?
- Présentation, avec des exemples concrets, de la démarche.
- Détermination du «juste nécessaire» pour assurer la disponibilité d’un système d’in-formation.
- Identification des facteurs déterminants pour la réussite d’un projet de sécurisation du système d’information : le DIC du CLUSIF et les thèmes ISO.
- Maîtriser les étapes, les concepts et les dé-marches de la sécurisation, les principales activités de sécurisation.
2.3. Justifier et obtenir les moyens de la politique sécurité grâce à l’Analyse de Risques (AR) - Définition et présentation des objectifs de l’Analyse de Risques.
- Rappel des concepts de base et de la théorie de l’analyse de risque.
- Modèle métier et sensibilité des métiers (BIA Business Impact Analysis), le recueil des informations.
- Identification des dangers et des menaces, notamment des causes de défaillance tech-nique (HI Hazard Identification).
- Analyse de la vulnérabilité : étude des impacts et des moyens de supprimer les menaces techniques ou de limiter les impacts métiers (HA Hazard Analysis).
- Détermination des enjeux de la politique sécurité : Valorisation des impacts potentiels (Business Impact Valuation).
2.4. La méthodologie et les technologies de la sécurité - Comment mettre en place une politique de sécurité efficace ?
- Comment intégrer les référentiels norma-tifs et réglementaires dans la politique de sécurité ?
- Comment utiliser les standards du domaine afin de construire une politique de sécurité efficace ?
- Quels sont les outils qui permettent d’inté-
grer les différentes composantes ? EBIOS, MEHARI, …
- Qu’est-ce qu’un Système de Management de la Sécurité Informatique (SMSI) ?
- Comment définir et mettre en œuvre un SMSI ?
3. La sensibilisation / la formation3.1 La formation - Quelle est la différence entre la sensibilisa-tion et la formation ?
- A qui s’applique la formation ?3.2 La sensibilisation - Comment faire pour sensibiliser les utilisa-teurs aux problèmes de sécurité ?
- Qu’est-ce qu’une charte informatique ? - Quels sont les moyens légaux pour faire adopter une charte ?
EXERCICES DE MISE EN SITUATIONUn cas pratique en forme de fil rouge sur le système d’information d’une entreprise est déroulé tout au long de cette formation ; les étapes concernées sont : - analyse de risque métier - analyse de risque infrastructure informa-tique
- politique sécurité - système de management de la sécurité informatique
- sensibilisation et formation
Un QCM vous est proposé afin d’évaluer les connaissances acquises.
Gestion de la sécurité des systèmes d’informations
C61NOUVEAU
C58
Pour développer vos compétences, nous vous conseillons également :
B9C60
C59
B9
Vous souhaitez obtenir
plus d’informations ?
Contactez-nous + 212 (0)6 61 13 69 28
www.2ssante.com

AUDITS / INSPECTIONS

AU
DIT
S /
INS
PE
CT
ION
S
43Pour toute formation intra-entreprise : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
L’audit fournisseur fait partie intégrante du processus de décision du choix d’un fournisseur, mais permet également d’adapter la stratégie de l’entreprise vis-à-vis d’un fournisseur imposé. Il permet d’apporter les garanties en qualité de conception, de production et de services.Les exercices proposés pour cette formation sont sous forme de jeux de rôle et ils permettent une réelle mise en application des méthodes enseignées.
1. Pourquoi auditer ?Définition d’un audit. Enjeux, objectifs et
périmètre. Terminologie associée. Activités
et acteurs concernés.
La relation client-fournisseur et les respon-
sabilités de chaque partie. Le contexte régle-
mentaire (BPF/cGMP, BPL/GLP, BPC/GCP, 21
CFR part 11, …).
2. Processus d’auditLes normes et guides applicables (ISO, GAMP,
PDA, …).
Le processus d’audit :
1 – Définir le périmètre de l’audit
2 – Organiser l’audit
3 – Préparer un questionnaire détaillé
4 – Réaliser l’audit sur place
5 – Préparer un rapport
6 – Décider et suivre
L’auditeur : critères, qualités personnelles,
contexte.
Prise de conscience de l’auditeur par l’audité
L’audit : aspects humains et culturels.
3. Principes de l’ingénierie et de la qualité informatiqueCycle de vie logiciel : processus de base, de
support, et organisationnel.
Responsabilités et Acteurs.
Gestion de la configuration
Gestion des modifications
4. Méthodes et référentiel de l’auditDomaines et points à inclure dans le question-
naire d’audit : développement, exploitation,
maintenance, sécurité, formation, gestion de
projet, documentation, gestion de configura-
tion, modifications, …
Aides à l’élaboration du questionnaire : réfé-
rentiels reconnus GAMP, PDA, CMM, SPICE ;
analyses de risques; analyses de la réglemen-
tation, conseils et astuces, …
Non-conformités fréquemment rencontrées.
Techniques à employer lors du déroulement
de l’audit.
EXERCICE DE MISE EN SITUATION
- Préparation de l’auditExercice sous forme d’un cas réel à traiter,
permettant une mise en application directe
des méthodes enseignées.
- Réalisation de l’auditJeux de rôles, en utilisant les questionnaires
précédemment préparés.
- Bilan de la mise en situationDiscussion des expériences lors de l’essai d’audit. Récapitulatif des points clés de l’audit.
Un QCM vous est proposé afin d’évaluer les
connaissances acquises.
Audit des fournisseurs de systèmes automatisés et informatisés
Objectifs
● Connaître les différentes étapes pour
réaliser un audit fournisseur.
● Connaître les exigences réglementaires
et normatives applicables pour réaliser
des audits pertinents et performants.
● Savoir définir l’objectif et justifier l’audit
fournisseur.
● Préparer le questionnaire d’audit en
fonction du référentiel choisi.
● Connaître les points d’audit les plus im-
portants.
● Savoir restituer les observations et/ou les
déviations observées au cours de l’audit
à travers le rapport.
Programme
A18
Formation inter-entreprises
DUREE 2 jours
PUBLIC
- Equipes d’audit fournisseur (AQ, Infor-
matique, Key User)
- Fournisseurs de l’industrie de la santé
susceptibles d’être audités
B103
Pour développer vos compétences, nous vous conseillons également :
B104
B104
B34
Toutes nos formations sont
disponibles en anglais
Contactez-nous + 212 (0)6 61 13 69 28
www.2ssante.com

44
AU
DIT
S /
INS
PE
CT
ION
S
Pour tout projet de formation : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
1. Exigences réglementaires et référentiels normatifsQuelles sont les normes liées aux audits-?
Présentation des différents référentiels de
l’assurance qualité réglementaire (BPF/
cGMP, BPL/GLP, BPC/GCP, ICH, …) Présen-
tation des référentiels normatifs généraux
et spécifiques à l’audit (ISO 9000:2005, ISO
9001:2008, ISO 19011:2002, ISO 100XX …).
2. Le système qualité et les outils qualité
La qualité, c’est quoi ? Description de la po-
litique qualité, du rôle du management de
la qualité et des outils nécessaires (manuel
qualité, plan qualité, procédures, mode opé-
ratoire, …) et de la gestion documentaire.
3. Audits - DéfinitionsQuels termes utilise-t-on dans le domaine de
l’audit et que signifient-ils ? Définition des
termes relatifs à la documentation qualité
(manuel qualité, procédures, processus, …) et
ceux relatifs à l’audit (revue, audit, inspection,
rapport, exigences, auditeurs, audités, …).
Appréhender l’ensemble des mots clés de
l’audit correspondant à la méthodologie,
aux principes, à la chronologie et à la notion
d’indépendance.
4. Audits - Aspects culturels et humainsQuels sont les aspects culturels et humains à
prendre en compte ? Le mot Audit vient du
latin « Audire » qui signifie « Ecouter » ! L’au-
diteur doit prendre compte les composantes
humaines et culturelles (stress, objectivité,
confidentialité de l’audité, …) tout en conser-
vant son indépendance et sa neutralité.
5. Audits - Objectifs, missions, techniques et attitudesQuels sont les objectifs de l’audit ? Ce n’est pas
seulement de lister des problèmes ! Quelles
sont les missions de l’audit ? L’audit est un ou-
til commun au service de tous ! Quelles sont
les techniques liées à l’audit ? La grille d’audit
n’est pas une finalité mais un point de départ-!
Quelles sont les attitudes liées à l’audit ? Un
auditeur est humain avec ses forces et ses fai-
blesses ! Connaître les différentes techniques
d’audit par l’observation, par des questions
ouvertes ou fermées, par étude de documents,
par entretiens, …, avec leurs avantages et leurs
inconvénients.
6. Audits - Déroulement de A à ZConnaître les différentes phases de l’audit,
de la préparation avec son périmètre d’appli-
cation et son objectif à la notification avec la
planification puis à sa réalisation. Préparer la
réunion de lancement avec la présentation de
l’objectif.
Restitution et conclusion de l’audit (points
forts, points faibles et observations, …). Ré-
daction du rapport d’audit ; un écart n’est pas
forcément une observation ! Une observation
doit être supportée par des faits (preuves) !
Suivi des actions correctives.
7. Indicateurs et tableaux de bordsDéfinir le contenu d’un tableau de bord avec
des indicateurs pertinents, qualifiables et
quantifiables permettant de suivre la confor-
mité et l’aptitude à satisfaire aux exigences et
à corriger les non-conformités.
L’audit qualité interne doit aussi et surtout être un moyen de communication dans l’entreprise.L’audit qualité interne fait partie intégrante du système qualité ; il est en quelque sorte l’assurance qualité du système qualité. Quand il est correctement utilisé, c’est un vrai moyen d’amélioration continue de la qualité et de maintien ou de mise en conformité réglementaire. Au travers du rapport d’audit et du suivi des actions correctives, l’évolution du système qualité sera garantie. L’audit qualité interne vérifiera aussi la pertinence et l’efficacité des indicateurs qualité pour la maîtrise des processus et le suivi des tableaux de bords.
8. Revue de l’ISO 19011:2002Revue des recommandations contenues
dans la norme.
9. Etude de casSimulation d’un audit avec auditeurs et au-
dités : Audit d’un processus d’audit qualité
interne.
Sélection des référentiels applicables,
Confrontation du contenu de la procédure
objet de l’audit aux exigences définies, Pré-
paration d’un questionnaire d’audit, Simula-
tion du déroulement de l’audit (auditeurs /
audités) : expression d’observations, apport
de preuves, Analyse des faits et des écarts et
synthèse sous forme d’observation. Proposi-
tion de solutions de mise en conformité et
d’amélioration.
EXERCICES DE MISE EN SITUATIONPlusieurs exercices sont proposés pendant la
formation pour illustrer les points abordés.
Un QCM vous est proposé afin d’évaluer les
connaissances acquises.
Audit Qualité Interne - L’outil d’amélioration et de mise en conformité réglementaire (ISO 19011)
Objectifs
● Connaître le vocabulaire de l’assurance
qualité et plus particulièrement de l’au-
dit qualité.
● Comprendre les exigences réglemen-
taires et normatives applicables au sys-
tème qualité.
● Connaître les enjeux et les techniques de
l’audit interne.
● Connaître le déroulement de l’audit de
A à Z.
● Comprendre le rôle des indicateurs qua-
lité et des tableaux de bords qualité.
● Connaître les attitudes à adopter par les
auditeurs.
Programme
B103
Formation inter-entreprises
DUREE 2 jours
PUBLIC
- Assurance Qualité
- Production
- Contrôle Qualité
- Informatique
- Recherche & développement
- Ingénierie et automatisme
- Distribution
A18
Pour développer vos compétences, nous vous conseillons également :
B104
B10

45
www.cyberconseil.com
FRANCEBELGIQUE
SUISSEALLEMAGNE
ETATS-UNISCVO-CyberConseilLe partenaire des Industries de la Santé
• Production : formes sèches, liquides, injectables, équipements, utilités
• Informatique : systèmes informatisés, GED, LIMS, ERP (SAP®), Infrastructure Informatique
• Informatique industrielle / automatique : systèmes MES, supervision, …
• Laboratoire : R&D, Contrôle Qualité, équipements, …
• Procédés : NEP, SEP, décontamination, logistique, méthodes industrielles, …
Avec CVO-CyberConseil, assurez la réussite de vos projets :
CVO-CyberConseilLe partenaire des Industries de la Santé
Conseil et AssistanceINGÉNIERIE
QUALITÉVALIDATION
Depuis 1995, le groupe CVO-CyberConseil propose des solutions
en conseil et ingénierie aux Industries de la Santé. L’expertise de
ses consultants, ses méthodes innovantes, sa parfaite maîtrise des
réglementations applicables et des spécificités du secteur en font
un partenaire privilégié des Industries de la Santé.
CVO-CyberConseil intervient dans les secteurs suivants :
Pharmacie - Chimie - Cosmétique - Biotechnologies
Dispositifs Médicaux - Etablissements de Santé
Agro-alimentaire - Laboratoires / CRO

46
AU
DIT
S /
INS
PE
CT
ION
S
Pour tout projet de formation : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
L’Audit : Outil d’amélioration et de mise en conformité réglementaire (ISO 19011).L’audit qualité interne fait partie intégrante du système qualité ; il est en quelque sorte l’assurance qualité du système qualité. Quand il est correctement utilisé, c’est un vrai moyen d’amélioration continue de la qualité et de maintien ou de mise en conformité régle-mentaire. Au travers du rapport d’audit et du suivi des actions correctives, l’évolution du système qualité sera garantie. L’audit qualité interne vérifiera aussi la pertinence et l’efficacité des indicateurs qualité pour la maîtrise des processus. L’audit fournisseur fait partie intégrante du processus de décision du choix d’un fournisseur, mais permet également d’adapter la stratégie de l’entreprise vis-à-vis d’un fournisseur imposé. Il permet d’apporter les garanties en qualité de conception, de production, de systèmes, de produits, de matières premières et de services. Il permet aussi de répondre aux exigences réglementaires qui imposent ces audits.
1. Exigences réglementaires et référentiels normatifs liés à l’auditQuelles sont les normes liées aux audits-? Présentation des différents référentiels de l’assurance qualité réglementaire (BPF/cGMP, BPL/GLP, BPC/GCP, ICH, ISO, CFR …). Présentation des référentiels normatifs géné-raux à la qualité et à l’audit (ISO 9000:2005, ISO 9001:2008, ISO 19011:2002, ISO 100XX …)
2. Le système qualité et les outils qualité
La qualité, c’est quoi ? Description de la po-litique qualité, du rôle du management de la qualité et des outils nécessaires (manuel qualité, plan qualité, procédures, mode opé-ratoire, …) et de la gestion documentaire.
3. Pourquoi et qui auditer ?Activités et acteurs concernés. La relation client-fournisseur en interne et en externe et les responsabilités de chaque partie.Les différents types de fournisseurs : API, Sys-tèmes, Services …Quels fournisseurs auditer ?
4. Audits - DéfinitionsQuels termes utilise-t-on dans le domaine de l’audit et que signifient-ils ? Définition des termes relatifs à la documentation qualité (manuel qualité, procédures, processus, …) et ceux relatifs à l’audit (revue, audit, inspection, rapport, exigences, auditeurs, audités, …). Appréhender l’ensemble des mots clés de l’audit correspondant à la méthodologie, aux principes, à la chronologie et à la notion d’in-dépendance.
5. Audits - Aspects culturels et humainsQuels sont les aspects culturels et humains à prendre en compte ? Le mot Audit vient du latin « Audire » qui signifie « Ecouter » ! L’auditeur doit prendre en compte les com-posantes humaines et culturelles (stress, objectivité, confidentialité de l’audité, …) tout en conservant son indépendance et sa neutralité.
6. Audits - Objectifs, missions, techniques et attitudesQuels sont les objectifs de l’audit ? Ce n’est pas seulement de lister des problèmes ! Quelles sont les missions de l’audit ? L’audit est un outil commun au service de tous-!
Audit Qualité Interne et Audits Fournisseurs
Objectifs
● Connaître le vocabulaire de l’assurance qualité et plus particulièrement de l’audit qualité.
● Comprendre les exigences réglementaires et normatives applicables au système qualité.
● Connaître les exigences réglementaires et normatives applicables pour réaliser des audits pertinents et performants tant en interne que chez les fournisseurs.
● Connaître les enjeux et les techniques de l’audit interne.
● Connaître les différentes étapes pour réaliser un audit
● Connaître le déroulement de l’audit de A à Z.
● Comprendre le rôle des indicateurs qualité et des tableaux de bords qualité.
● Connaître les attitudes à adopter par les auditeurs.
● Connaître les points d’audit les plus im-portants.
● Savoir définir l’objectif et justifier l’audit fournisseur.
● Savoir préparer un audit et un question-naire / check liste approprié.
● Savoir réaliser la synthèse d’un audit et la formaliser dans un rapport clair et concis.
Programme
B104NOUVEAU
Formation inter-entreprises
DUREE 2 jours
PUBLIC
- Equipes d’audit fournisseur (AQ,
Matières premières, Services, Informa-
tique, Key User)
- Fournisseurs de l’industrie de la santé
susceptibles d’être audités
- Assurance Qualité
- Production
- Contrôle Qualité
- Informatique
- Recherche & Développement
- Ingénierie et Automatisme
- Distribution
B10

AU
DIT
S /
INS
PE
CT
ION
S
47Pour toute formation intra-entreprise : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
Quelles sont les techniques liées à l’audit ? La grille d’audit n’est pas une finalité mais un point de départ ! Quelles sont les attitudes liées à l’audit ? Un auditeur est humain avec ses forces et ses faiblesses ! Connaître les dif-férentes techniques d’audit par l’observation, par des questions ouvertes ou fermées, par étude de documents, par entretiens, …, avec leurs avantages et leurs inconvénients.
7. Audits - Déroulement de A à ZConnaître les différentes phases de l’audit, de la préparation avec son périmètre d’appli-cation et son objectif à la notification avec la planification puis à sa réalisation. Préparer la réunion de lancement avec la présentation de l’objectif.Restitution et conclusion de l’audit (points forts, points faibles et observations, …). Rédaction du rapport d’audit ; un écart n’est pas forcément une observation ! Une ob-servation doit être supportée par des faits (preuves) ! Suivi des actions correctives.
8. Indicateurs et tableaux de bordsDéfinir le contenu d’un tableau de bord avec des indicateurs pertinents, qualifiables et quantifiables permettant de suivre la confor-mité et l’aptitude à satisfaire aux exigences et à corriger les non-conformités.
9. Revue de l’ISO 19011:2002Revue des recommandations contenues dans la norme.
10. Référentiels réglementaires et norma-tifs liés aux audits fournisseursPrésentation des référentiels réglementaires et normatifs spécifiques aux audits des four-nisseurs (ISO, GAMP, ICH, CMM, SPICE …).Les différentes approches en fonction du type de fournisseurs. Aides à l’élaboration du questionnaire : analyses de risques; analyse des réglementations, analyse des référentiels, analyse des audits précédents, conseils et as-tuces, …
11. Spécificités des fournisseurs de sys-tèmes informatisés / automatisésPrincipes de l’ingénierie et de la qualité infor-matique : - Cycle de vie logiciel : processus de base, de support, et organisationnel
- Responsabilités et Acteurs - Gestion de la configuration - Gestion des modifications
Domaines et points à inclure dans le ques-tionnaire d’audit de fournisseurs de systèmes informatisés / automatisés : développement, exploitation, maintenance, sécurité, for-
mation, gestion de projet, documentation, gestion de configuration, modifications, …Non-conformités fréquemment rencontrées. Techniques à employer lors du déroulement de l’audit.
12. Etudes de cas : Simulation d’audits avec auditeurs et audités Etude de cas 1 : Audit d’un processus d’audit qualité interne.Sélection des référentiels applicables.Confrontation du contenu de la procédure objet de l’audit aux exigences définies.Préparation d’un questionnaire d’audit.Simulation du déroulement de l’audit (audi-teurs / audités) : expression d’observations, preuves.Analyse des faits et des écarts et synthèse sous forme d’observation.Proposition de solutions de mise en confor-mité et d’amélioration.Etude de cas 2 : Audit d’un fournisseur de matières premières.Sélection des référentiels applicables.Préparation d’un questionnaire d’audit.Simulation du déroulement de l’audit (audi-teurs / audités) : expression d’observations, preuves.Analyse des faits et des écarts et synthèse sous forme d’observation.Proposition de solutions de mise en confor-mité et d’amélioration.Etude de cas 3 : Audit d’un fournisseur de systèmes informatisés / automatisés.
Sélection des référentiels applicables.
Préparation d’un questionnaire d’audit.
Simulation du déroulement de l’audit (audi-
teurs / audités) : expression d’observations,
preuves.
Analyse des faits et des écarts et synthèse
sous forme d’observation.
Proposition de solutions de mise en confor-
mité et d’amélioration.
Un QCM vous est proposé afin d’évaluer les
connaissances acquises.
Les supports d’exercices sont fournis à
chaque participant, y compris les textes ré-
glementaires utilisés pour les exercices (BPF,
21 CFR, ICH …) et des extraits des textes
normatifs (ISO 19011, ISO 9000 …) dans la
limite du respect des copyrights.
Des exemples de plan d’audit, de questionnaire
/ check liste d’audit et de rapport d’audit sont
également fournis aux participants à la fin de la
formation.
Audit Qualité Interne et Audits FournisseursB104
NOUVEAU
A18
Pour développer vos compétences, nous vous conseillons également :
B103
A18
Toutes nos formations sont
réalisables en intra-entreprise
Contactez-nous + 212 (0)6 61 13 69 28
www.2ssante.com

48
AU
DIT
S /
INS
PE
CT
ION
S
Pour tout projet de formation : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
Les conséquences d’une investigation FDA ratée peuvent être tellement catastrophiques que l’annonce d’une inspection FDA génère un stress important. Comprendre la FDA, son rôle, sa mission et la façon de penser des investigateurs et des compliance officers permet d’être mieux préparé et d’aborder plus sereinement ces inspections. Réussir une inspection FDA tient souvent à peu de chose, tout le problème étant de savoir ce qui est important, ce qu’il faut dire, ce qu’il ne faut pas dire, comment répondre à une observation, quoi montrer, ... Avec une approche pratique, cette formation vous prépare à maîtriser votre relation avec la FDA et les autorités américaines.
Ce que les inspecteurs peuvent faire et ce qu’ils ne peuvent pas faire.Front room et Back room : a quoi ça sert et comment ça se gère ?A Faire et Ne Pas Faire (DOs & DON’Ts) pendant l’inspection.Que faire lorsque l’on est confronté à une question hypothétique ?Que faire lorsque l’on est confronté à une question quantitative ?Les mots ambigus à ne pas utiliser.Suite à une Warning Letter / 483 – Qu’est-ce qu’il faut faire ?
EXERCICES DE MISE EN SITUATIONExercices et études de cas sont proposés : - Analyse des notes d’un inspecteur suite à une inspection : Qu’est-ce qui ne va pas ? Qu’est ce que vous auriez dit à leur place ? Que proposeriez-vous comme réponse ?
- Analyse d’observations extraites de 483 : Quel est le problème ? Comment aurait-on pu l’éviter ? Que faut-il répondre ?
- Analyse d’observations extraites de Warning Letters : Quel est le problème ? Comment aurait-on pu l’éviter ? Que faut-il répondre ?
Un QCM vous est proposé afin d’évaluer les connaissances acquises.
1. Définitions et objectifs de la FDA Position de l’agence et son importance.Introduction à la FDA.FDA, sa mission, son rôle et son approche de l’industrie.Historique de la FDA.Qui est inspecté par la FDA ?Les notions de produits Purs, Surs et Efficaces.
2. Référentiels réglementaires et normatifs de la FDAFDA et ses organisations : (CBER, CDER, CVM, CFSAN, CDRH, NCTR and ORA).21CFR : Preamble, Subparts (21CFR 210/211, 21CFR 820 …).FDA Guidelines, FDA Inspection Guides, FDA Compliance Programs, FDA Policy Statements, Other Publications, Advisory Opinions.C’est quoi un produit « adultered » et ça a quelles conséquences ?Le manuel des opérations d’investigation de la FDA : Contenu et importance.Comment trouver la bonne information, et comprendre ce qui est explicite et implicite dans les 21CFR ?Comment trouver plus d’informations ?Comment la loi devient la loi ?
3. Les conséquences et le coût de la sou-mission à la FDAFDA – Freedom of Information Act – Le droit de savoir (liberté de l’information).Les conséquences et le « coût » d’une inspec-tion FDA.Les rappels de lot et la « MedWatch ».Excuses classiques !483 et Warning Letters.
4. Les différences entre les inspections Européennes et les investigations Améri-caines482, 483, EIR, Warning Letter ?Types et styles des inspections FDA : PAI, QSIT, « Risk-Based » (basée sur les risques).Différences par rapport aux inspections à l’étranger.cGMP et la FDA : le concept du progrès.Différences entre l’approche FDA et l’ap-proche Européenne et les conséquences de ces différences.Différences culturelles entre les USA et l’Eu-rope : Pourquoi des choses qui paraissent mineures pour les Européens peuvent être majeures pour les Américains ?
5. Comment préparer l’entreprise pour le futur par rapport à ce qui est désormais important dans l’industrieUne revue brève sur les “Hot Topics” (contre-façon, approche des risques, les médicaments «on line», l’impact du terrorisme, Critical Path, PAT) .Risk-Based Approach – Quoi ? Pourquoi ? Comment ? D’où ça vient ? Pourquoi main-tenant ? FDA - Le coût de la qualité.Bonne Pratiques Documentaires selon la FDA.
6. Le déroulement de l’inspection FDA. Comment être préparé, proactif, profes-sionnel ?Gérer les inspections FDA (avant, pendant, après).Qu’est ce qui va être inspecté ?Ce qu’il faut faire avant les inspections.
La FDA : missions, réglementations, préparationet déroulement des inspections
Objectifs
● Avoir tous les éléments pour une com-préhension pratique et fondamentale concernant la FDA.
● Comprendre les principes derrière les cGMPs et leur constante évolution.
● Connaître les exigences réglementaires, l’approche et les pratiques de l’industrie vis-à-vis des cGMPs.
● Mieux comprendre la structure de la FDA (ORA/CBER/CDER/CDRH/…) et ses objectifs.
● Connaître les différences culturelles
entre l’Europe et les Etats-Unis et leurs influences sur le déroulement des inspec-tions et investigations.
● Savoir comment gérer une inspection FDA avant, pendant et après (Front room, Back Room …).
● Savoir communiquer de façon efficace avec la FDA (les « Dos » et les « Don’ts »).
● Comprendre pourquoi la FDA met tou-jours la barre plus haut.
● Découvrir les sujets courants et nouveaux (« hot topics ») et anticiper les futures évo-lutions de la FDA.
Programme
B8
FORMATION ACTUALISEE
Formation inter-entreprises
DUREE 2 jours
PUBLIC
- Assurance et Contrôle Qualité
- Affaires Réglementaires
- Production
- Maintenance/Métrologie
- Recherche et Développement
- Pharmacovigilance
- Essais cliniques
- Informatique
- Qualification/Validation
- Management projet
B30

AU
DIT
S /
INS
PE
CT
ION
S
49Pour toute formation intra-entreprise : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
1. Définitions et objectifs du MHLW et du PMDA. Position de l’agence et son importance.
Introduction au MHLW.
Le MHLW, sa mission, son rôle et son approche
de l’industrie.
Historique du MHLW.
Introduction au PMDA.
Le PMDA, sa mission, son rôle et son approche
de l’industrie.
Historique du PMDA.
Qui est inspecté par le MHLW ou le PMDA ?
2. Référentiels réglementaires et normatifs du MHLW. Le MHLW et ses organisations.
Le PMDA et ses organisations.
Good Manufacturing Practices - GMP, Good Vi-
gilance Practices - GVP, Good Quality Practices
– GQP, Quality Management System - QMS.
Le MHLW, le PMDA et les ICH.
Comment trouver la bonne information, et
comprendre ce qui est explicite et implicite
dans les textes de loi ?
Comment trouver plus d’informations ?
3. Les conséquences et le coût de la sou-mission au MHLW.Les conséquences et le « coût » d’une inspec-
tion Japonaise.
Les rappels de lot et la Vigilance.
Observations et lettre d’avertissement.
4. Les différences entre les inspections Eu-ropéennes et les inspections JaponaisesTypes et styles des inspections.
MHLW, PMDA et le concept d’assurance qualité.
Différences entre l’approche Européenne et
l’approche Japonaise et les conséquences de
ces différences.
Différences culturelles entre le Japon et l’Eu-
rope : Pourquoi des choses qui paraissent
mineures pour les Européens peuvent être
majeures pour les Japonais ?
5. Comment préparer l’entreprise pour le futur par rapport à ce qui est désormais important dans l’industrieUne revue brève sur les “Hot Topics” .
Risk-Based Approach – Quoi ? Pourquoi ?
Comment ? D’où ça vient ? Pourquoi main-
tenant ?
Bonnes Pratiques Qualité et ICH Q10 : Les Ja-
ponais sont en avance !
Bonne Pratiques Documentaires.
6. Le déroulement de l’inspection. Com-ment être préparé, proactif, professionnel ?Gérer les inspections Japonaises (avant, pen-
dant, après).
Qu’est ce qui va être inspecté ?
Ce qu’il faut faire avant les inspections.
Ce que les inspecteurs peuvent faire et ce
qu’ils ne peuvent pas faire.
Front room et Back room : a quoi ça sert et
comment ça se gère ?
En 2006, une nouvelle loi « Pharmaceutical Affairs Law - PAL » du ministère de la santé Japonais qui impose que toutes les sociétés qui exportent des produits pharmaceutiques au Japon soient auditées par les instances Japonaises a été adoptée. Ceci a pour effet, depuis 2009, qu’il y a de plus en plus d’inspections Japonaises en Europe. Les conséquences d’une inspection Japonaise ratée peuvent être catastrophiques vu l’ampleur du marché. Comprendre l’organisation des instances réglementaires Japonaises, leur rôle, leur mission et la façon de penser des inspecteurs Japonais permet d’être mieux préparé et d’aborder plus sereinement ces inspections. Dès l’instant que l’on travaille correctement, réussir une inspection tient souvent à peu de chose, tout le problème étant de savoir ce qui est important, ce qu’il faut dire, ce qu’il ne faut pas dire, comment répondre à une observation, quoi montrer, ... Avec une approche pratique, cette formation vous prépare à maîtriser votre relation avec les autorités Japonaises.
A Faire et Ne Pas Faire (DOs & DON’Ts) pendant
l’inspection.
Suite à une Observation – Qu’est-ce qu’il faut
faire ?
EXERCICES DE MISE EN SITUATIONExercices et études de cas sont proposés : - Simulation d’une inspection : sur un sujet proposé par le formateur ou sur un sujet proposé par les participants.
- Analyse d’observations : Quel est le pro-blème ? Comment aurait-on pu l’éviter ?
Que faut-il répondre ?
Un QCM vous est proposé afin d’évaluer les
connaissances acquises.
Les inspections Japonaises, le MHLW et le PMDA
Objectifs
● Avoir tous les éléments pour une com-
préhension pratique et fondamentale
concernant le Ministry of Health, Labour
and Welfare – MHLW et le Pharmaceuti-
cals and Medical Devices Agency - PMDA.
● Mieux comprendre la structure du MHLW
et du PMDA et leurs objectifs.
● Connaître les différences culturelles entre
l’Europe et le Japon et leurs influences sur
le déroulement des inspections.
● Savoir comment gérer une inspection
Japonaise.
● Savoir communiquer de façon efficace
avec le MHLW et le PMDA (les « Dos » et
les « Don’ts »).
● Comprendre pourquoi le MHLW et le
PMDA sont très sensibles à la Qualité.
Programme
B35NOUVEAU
Formation inter-entreprises
DUREE 2 jours
PUBLIC
Toute personne qui peut être confrontée
à une inspection des autorités Japo-
naises.
B30
B10

INGÉNIERIE / PROJETS

ING
EN
IER
IE /
PR
OJE
TS
51Pour toute formation intra-entreprise : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
1. IntroductionDéfinitions. Objectifs de la revue de concep-tion. Cycles de vie et types de projet. Comment agir dans une incertitude qui décroît. Revue de conception et GAMP. Vérification versus Validation.
2. Référentiels réglementaires et normatifs
Description des référentiels applicables. Revue de la position et des attentes des ins-tances réglementaires en termes de Revue de conception.
3. Documentation des revues de concep-tionComment documenter une revue de conception ? Eléments d’entrée de la concep-tion et du développement. Eléments de sor-tie de la conception et du développement. Vérification de la conception et du dévelo-ppement. Validation de la conception et du développement. Maîtrise des modifications de la conception et du développement. Doit-on écrire des protocoles, utiliser des listes de vérification (check-list). Qui participe aux re-vues de conception. Que doit-on documenter.
4. RC des Spécifications des Besoins Utili-sateurs (Cahier des Charges - CdC)Présentation des méthodes, outils et tech-niques de revue des Cahiers des Charges, qui permettent de valider les besoins compte tenu des objectifs. Définitions : Cahier des
charges, Spécifications, Exigences,…Cahier des charges et cycle de vie projet. Méthodo-logie d’élaboration d’un cahier des charges. Comment rendre les objectifs SMART ?
5. RC des Spécifications FonctionnellesPrésentation des méthodes, outils et tech-niques de revue des Spécifications Fonction-nelles, qui permettent de valider les choix fonctionnels. Vérifier la traçabilité des exi-gences.
6. RC des Spécifications Techniques et du Dossier de ConceptionPrésentation des méthodes, outils et tech-niques de revue de la conception du sys-tème et des Spécifications Techniques, qui permettent notamment de valider les choix techniques. Documentation nécessaire. Les fiches de vie.
7. RC des Programmes et LogicielsPrésentation des méthodes, outils et tech-niques de revue des programmes et des logiciels. Qu’est ce qu’une revue de code ? Comment vérifier que de bonnes pratiques de programmations ont été suivies.
8. RC des TestsPrésentation des méthodes, outils et tech-niques de revue des tests. Les différents types de tests. Tests fournisseurs versus tests utili-sateurs.
Revue de Conception, Design Review, Qualification de Développement, Design Qualification Appelée également DQ (Design Qualification), QD (Qualification de Développement), RCE (revue de Conception Etendue), EDR (Enhanced
Design Review), cette étape de vérification garantit que toutes les exigences réglementaires et business, sont non seulement prises en compte
dès le début du projet, mais aussi qu’elles sont correctement transcrites et mises en œuvre tout au long du cycle de développement, sans
oublier la génération des codes source. Elle permet de détecter les défauts au plus tôt et de les corriger au moindre coût, et ainsi d’éviter les
dérives budgétaires et de planning. Réalisée sur des systèmes déjà en production, elle permet d’évaluer le niveau de conformité réglementaire
du système et de sa documentation associée afin de définir un plan d’action de mise en conformité adapté.
RC / QD : Revue de Conception / Qualification de Développement
Objectifs
● Définir des stratégies de revue adaptée au contexte projet (maturité des fournisseurs, bonnes pratiques…).
● Connaître les exigences réglementaires et normatives applicables aux Revues de Conception et déterminer les informa-tions du dossier de Revue de Conception indispensables pour être en conformité avec les exigences réglementaires de l’AFSSAPS, de l’EMEA, de la FDA (BPF/GMP, BPL/GLP, BPC/GCP, QSR…), de l’ISO (9001, 13485), ...
● Savoir définir et optimiser l’effort de revue nécessaire en fonction du projet et répartir
les activités de revue entre les différents acteurs.
● Savoir faire la relation entre Revue de Conception et Analyse de Risques.
● Etre capable de définir et mettre en œuvre les Revues de Conception Etendue (RCE / EDR).
● Connaître l’étendue des techniques nécessaires pour réussir les Revues de Conception des systèmes industriels.
● Exploiter les résultats de la revue dans le reste du dossier de validation.
Programme
9. Relation entre Audit fournisseur et RC Comment utiliser l’audit fournisseur dans le cadre des revues de conception. L’audit four-nisseur, le pré-requis à la validation intégrée.
10. Relation entre Analyse de Risques et RC Comment utiliser les Analyse de Risques dans le processus de Revue de Conception. Rappels sur la gestion des risques (ISO 14971 / ICH Q9).
EXERCICES DE MISE EN SITUATIONPlusieurs exercices sont proposés pendant la formation pour illustrer les points abordés.Un QCM vous est proposé afin d’évaluer les connaissances acquises.
C20
A17
Formation inter-entreprises
DUREE 2 jours
PUBLIC
- Ingénierie, bureau d’études
- Informatique Industrielle et automa-
tisme (projets)
- Informatique IS/IT (projets)
- Maintenance / Métrologie
- Production / Exploitation
- Tests / Qualification / Validation
- Développement industriel / procédés
- QSE Qualité Sécurité Environnement
DOMAINES COUVERTS
- mécanique
- logiciel
- automatique
- électrique
B34A18
B9

52
ING
EN
IER
IE /
PR
OJE
TS
Pour tout projet de formation : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
Bien choisies et bien utilisées les statistiques sont des outils précieux en validation, pour confirmer ou infirmer des hypothèses, aider à démontrer l’aptitude des systèmes, ainsi que la robustesse des procédés. Il est d’ailleurs très difficile, voire impossible, de démontrer la capabilité d’un équipement, la reproductibilité d’un processus ou de faire de l’échantillonnage sans utiliser les statistiques. Vérifier l’équivalence de deux ou plusieurs populations peut aussi s’avérer utile mais parfois difficile sans un mini-mum de connaissances statistiques. Cette formation vous apportera les bases pour savoir dimensionner les principaux outils statistiques, les utiliser à bon escient et les justifier auprès des instances réglementaires.
1. Origine des statistiquesQuelle est l’efficacité d’un contrôle visuel à 100% réalisé par une personne ? Est-ce qu’il permet de détecter 100% des dé-fauts-? Est-il plus efficace et moins coûteux de faire de l’échantillonnage ? Rappel des généralités sur les probabilités. Relation entre population, nombre de défectueux, nombre d’échantillons.Calcul des probabilités. Les principes de bases sont présentés à l’aide d’exemples lu-diques basés sur des jeux de cartes, des dés, et le très connu pile ou face.
2. Les principales distributionsMoyenne. Variance.Exercice 1 – Calculs de variancesEcart type.Exercice 2 – Calculs d’écarts typeDistribution Normale de Gauss. Distribution Normale Centrée Réduite.Exercice 3 – Vérification de la normalité avec un histogrammeDistribution du Khi2. Distribution du Binôme. Distribution Hypergéométrique. Distribution de Poisson.
3. Statistiques utilisées en mise au point, validation et contrôle en cours de fabrica-tionPrésentation du rôle de la mise au point, de la validation et du contrôle en cours de fabrica-tion. Liens entre mise au point, validation et
contrôle en cours de fabrication. Présentation des principales statistiques utilisées en mise au point, validation et contrôle en cours de fabrication.Quand utiliser les statistiques ?
4. CapabilitéLes limites du raisonnement traditionnel.Pré-requis aux calculs de capabilité. Relation entre capabilité et tolérance. Quels sont les indicateurs qui permettent de déterminer la capabilité d’un processus ? Quelles sont les différences entre Cp, Cpk, Pp, Ppk ?Exercice 4 – Calculs de Cp/Pp et Cpk/PpkComment utiliser la capabilité pour détermi-ner le pourcentage probable de défectueux ?Exercice 5 – Calculs de pourcentage de défectueux à partir des Cp et CpkComment la notion d’écart de confiance intervient-elle sur la capabilité ? Les indica-teurs Cpm et Ppm.
5. Plan d’échantillonnage et carte de contrôleQu’est ce qu’un plan d’échantillonnage pour libérer des lots ? Quelles informations sont nécessaires pour déterminer le nombre d’élé-ments à prélever à chaque échantillon pour un plan basé sur l’ISO 2859 ? Définitions de AQL / NQA. Comment déterminer le NQA ? Relations entre NQA, Analyse de Risques, Tolérances, Capabilité. Définition et détermi-nation des coefficients Alpha et Bêta. Qu’est ce qu’une C-Curve (Courbe d’efficacité) ?
Initiation aux Statistiques utilisées en validation, contrôles et tests
Objectifs
● Connaître les termes utilisés en statis-tiques et leur signification.
● Connaître les principales distributions : Normale de Laplace-Gauss, Normale Cen-trée Réduite, du Khi2, du Binôme, Hyper-géométrique, de Poisson, de Student.
● Savoir utiliser les statistiques liées à une distribution Normale (Courbe de Gauss), Hypergéométrique, Binomiale, …
● Savoir construire un histogramme.
● Connaître les différentes statistiques utilisables en mise au point, validation et contrôle en cours de fabrication.
● Savoir construire et utiliser les cartes de contrôle.
● Savoir faire le lien entre Analyse des Risques, Niveau de Qualité Acceptable (NQA) et Capabilité.
● Savoir calculer et utiliser les capabilités (Cp, Cpk …).
● Connaître les pré-requis pour déterminer un plan d’échantillonnage.
● Savoir construire un plan d’échantillon-nage basé sur l’ISO 2859.
● Savoir déterminer et justifier le nombre d’échantillons prélevés.
● Connaître les principaux tests sur les va-riances et moyennes et leur utilisation.
Programme
Exercice 6 – Détermination d’un plan d’échantillonnage pour libérer des lotsPlan d’échantillonnage basé sur la distribu-tion du binôme dans le cas particulier de 0 défaut.Quand l’utiliser ? Comment l’utiliser ? Qu’est ce qu’une carte de contrôle ? Comment ça se construit ? Comment ça s’utilise ?Exercice 7 – Construction d’une carte de contrôle
6. Présentation de quelques tests basés sur les statistiquesRégression linéaire. Concept d’hypothèse nulle et d’hypothèse alternative. Loi de Fisher Snédécor. Distribution de Fisher Snédécor. Test de Fisher Snédécor (F test). Comment comparer 2 Variances avec le F test ?Exercice 8 – Comparaison de variancesLoi de Student. Distribution de Student. Pré-requis au test de Student (t test). Test de Student (t test). Comment comparer une moyenne à une norme avec le t test ? Com-ment comparer 2 moyennes avec le t test ?Exercice 9 – Comparaison de moyennes
EXERCICES DE MISE EN SITUATIONPlusieurs exercices sont proposés pendant la formation pour illustrer les points abordés.
Un QCM vous est proposé afin d’évaluer les connaissances acquises.
A20
Formation inter-entreprises
FORMATION ACTUALISEE
DUREE 2 jours
PUBLIC
- Assurance Qualité
- Contrôle Qualité
- Développement industriel
- Maintenance / Métrologie
- Production
- Qualification / Validation

ING
EN
IER
IE /
PR
OJE
TS
53Pour toute formation intra-entreprise : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
GxP Manager est un logiciel innovant qui permet de gérer les données techniques et réglementaires pour un projet, un service, un site ou toute une entreprise grâce à ses configurations standards totalement paramétrables aux besoins et spécificités des utilisateurs. GxP Manager permet la gestion exhaustive et sécurisée de données partagées dans une base et structurées grâce à un puissant moteur de liens. Le déploiement des solutions métiers de GxP Manager apporte des gains de productivité sensibles et très rapidement mesurables.
Une base commune d’apprentissage de 2
jours permet d’appréhender les fonctions de
base de GxP Manager. Toutefois la durée et
le contenu du programme sont totalement
adaptables en fonction des Solutions Métiers
choisies par les utilisateurs.
Formation de base standard : - Gestion des données : toutes les fonctions
de base vous permettant de spécifier vos
éléments : (besoins utilisateurs, spécifica-
tions fonctionnelles, cas de Tests…), et en-
suite de les travailler (modifier, copier, trier,
filtrer, hiérarchiser…).
- Import des données : fonctions standards et
avancées pour intégrer automatiquement les
données déjà existantes dans GXP Manager.
- Gestion la réglementation : sélection des
textes réglementaires dans la base web
GxP Knowledge Repository (KR), import des
textes fragmentés dans le logiciel.
- Lien des données : utiliser le moteur de
liens (liens de traçabilité, liens de composi-
tion, liens d’application).
- Génération automatique des matrices de
traçabilité personnalisées.
- Publication des documents (Cahier des
charges, Protocoles, Fiches de tests, Plans
de validation, Rapports…) : configurer et
automatiser la publication des données
techniques directement dans MS-Word selon
toutes les spécificités des modèles d’un pro-
jet ou d’une entreprise.
- Effectuer des analyses de risques multicri-
tères selon des méthodes standards (CVO-
RM, AMDEC/FMEA, HACCP, HAZOP, …) ou
personnalisées.
- Exécuter des documents de manière
interactive : exécuter des fiches de tests, des
questionnaires d’audits et tout type de
document technique dont les données
sont spécifiées dans GxP Manager.
Formation configurateur :Création et modification de bases par le
paramétrage de GxP Manager et sans aucun
développement informatique, pour adapter
et architecturer au mieux votre base selon les
besoins particuliers.
GxP Manager : la gestion des données métiers, projets et réglementaires
Objectifs
● Savoir utiliser les différentes fonctions de GxP Manager à travers des Solutions Mé-tiers telles que :
- Gestion du cycle de vie projet avec ges-tion : de la réglementation, des phases d’ingénierie (besoins utilisateurs, spéci-fications fonctionnelles et techniques), des analyses de risques (de l’analyse de criticité aux analyses multicritères types amdec…) des tests et de la qualification (rédaction, rapport et exécution électro-nique), du change control et de la main-tenance, …
- Gestion Multi-projets et Activités Ser-vice/Site (VMP-Etat des Lieux, Transferts et Déménagements, Laboratoires, Ingé-nierie Bâtiments, Infrastructure Informa-tique…),
- Gestion Qualité et Conformité (Veille Réglementaire-Gestion Référentiels et Normes, CAPA-Déviations et Non-confor-mités, Change Control, Audits, Conformité HSE, ...).
● Savoir utiliser le configurateur de GxP Manager pour créer ou adapter des Solutions Métiers.
Programme
A30
FORMATION ACTUALISEE
DUREE
- Fonctionnalités standards : 2 jours - Configurateur : 1 jour
PUBLIC
- Managers Qualité
- Managers Ingénierie
- Managers Projets
- Responsable Informatique
- Laboratoires et leurs équipes
- Ingénieurs, assistants
- Consultants et correspondants
Téléphone : +33 (0) 426 100 810
Email: [email protected]
www.gxpmanager.com
GxPMANAGER

54
ING
EN
IER
IE /
PR
OJE
TS
Pour tout projet de formation : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
1. Enjeux de la qualité informatique - Savoir justifier la mise en œuvre des bonnes pratiques informatiques dans les industries de la santé.
- Connaître les exigences réglementaires ap-
plicables.
Bref historique de l’informatique. L’informa-
tique d’aujourd’hui et de demain. Risques
associés à l’informatique. Exigences
réglementaires et exigences économiques.
Synthèse des textes réglementaires appli-
cables dans les domaines de la production
pharmaceutique (BPF, GMP, ICH), de la R&D
pharmaceutique (BPL, BPC, GCP, GLP, ICH), des
dispositifs médicaux (Quality System Regu-
lation), et à l’ensemble des activités (21 CFR
Part 11).
2. Connaître les objectifs, le contenu et les mérites des référentiels de qualité et d’ingénierie informatiquePrincipes de la Qualité. Définition d’un sys-
tème qualité. Avantages, du point de vue
de l’Assurance Qualité réglementaire, des
services utilisateurs et du personnel infor-
matique. Définition d’un processus informa-
tique. Présentation des modèles de processus
informatiques. Présentation des référentiels :
ISO9000, IS012207, ISO15504 (SPICE), CMMI,
ITIL, IEEE, GAMP, FDA Software Development
Activities, COBIT. Structures documentaires
proposées. Points-clés à retenir.
3. Présentation de l’approche ITIL d’un Système Qualité InformatiquePrésentation générale d’ITIL. Les Sept (7) mo-
dules constitutifs d’ITIL. Rappel des enjeux
majeurs. Intégration du système qualité au
sein du service informatique. Définition des
objectifs de qualité de service. Analyse des
principaux processus : fourniture, exploi-
tation, sécurité, sauvegarde et archivage,
support utilisateur, maintenance, gestion
des incidents, change control, gestion des
standards bureautiques, gestion du parc
bureautique,...
4. Apport d’ITIL dans les missions de SQI et de Qualification d’InfrastructureConnaître comment les modules d’ITIL peu-
vent aider à la mise en place d’un SQI et à la
Qualification d’Infrastructure informatique.
Rappel du positionnement d’ITIL dans le sys-
tème qualité. La qualification d’infrastructure
et ITIL.
5. Description détaillée d’ITILPrésentation en profondeur des principaux
modules d’ITIL :
- Le Service Support et ses processus : les
activités quotidiennes de l’IT ; les objectifs
du service desk et les liens avec les autres
processus IT ; le processus de gestion des
incident ; le processus de gestion des
problèmes ; le processus d’identification
des problèmes ; le processus de gestion du
changement ; le processus d’implémentation
en production ; la gestion des configurations.
- Les processus du Service Delivery : définition
du SLM (Service Level Management) et ses
déclinaisons (SLA, OLA) ; le processus de
gestion de la disponibilité (Availability
Avec l’informatisation croissante des entreprises, leur bon fonctionnement ainsi que le respect des exigences réglementaires pharmaceutiques reposent de plus en plus sur la performance des services informatiques, tant au niveau des études qu’au sein des équipes d’exploitation. Cette formation présente l’état de l’art des bonnes pratiques d’informatique, permettant au stagiaire d’intégrer celles-ci au sein de ses propres activités, afin d’améliorer sa maîtrise des risques et de maintenir le niveau de service requis.
management) ; La gestion des capacités IT ;
le processus de gestion de la continuité de
service IT (mise en place d’un BCP et d’un
DRP) ; l’approche financière de la gestion
d’un service IT ;
- La gestion de l’infrastructure IT : évolution
de l’infrastructure ; les liens avec les autres
modules ITIL ; le processus de déploiement ;
processus d’exploitation ; le support tech-
nique ;
- Le Security Management : les points clefs ;
les mesures de sécurité ;
- Le processus de gestion des applications : les
diverses phases du cycle de vie d’une appli-
cation
6. ITIL et exigences règlementairesComment ITIL permet de répondre aux
exigences règlementaires françaises (BPF
– LDP5), européennes (GMP – annex 11)
et américaines (FDA ; 21 CFR 11 ; 21 CFR
210/211).
Un QCM vous est proposé afin d’évaluer les
connaissances acquises.
Qualité Informatique : en conformitéavec les recommandations de l’ITIL
Objectifs
● Savoir justifier la mise en œuvre d’une
démarche de qualité informatique dans
les Industries de la Santé.
● Connaître les objectifs, le contenu et
les mérites des référentiels applicables
(IS012207, CMMI, ITIL, SPICE,...).
● Savoir identifier les processus informa-
tiques à la fois au niveau des études et en
production.
● Connaître les bonnes pratiques à
implémenter afin d’assurer le respect des
exigences réglementaires et le bon fonc-
tionnement de l’outil informatique dans
l’entreprise.
● Utiliser ITIL afin de répondre aux exi-
gences règlementaires pharmaceutiques.
Programme
B14
PUBLIC
- Assurance Qualité
- Assurance Qualité IS / IT
- Informatique
- Informatique (Projet)
- Ingénierie et Automatique
- Qualification / validation

ING
EN
IER
IE /
PR
OJE
TS
55Pour toute formation intra-entreprise : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
1. Qu’est-ce que le GAMP ?Le GAMP, ce qu’il est, ce qu’il n’est pas. Pour-
quoi a-t-il été écrit et par qui ? Quels sont ses
objectifs ? Comment est-il reconnu aujourd’hui
dans le monde de la santé, par les industriels,
les SSII et par les autorités réglementaires-? Quelle est son périmètre d’application ?
Comment est-il structuré ?
2. Les principes fondateurs du GAMPComprendre pourquoi et comment le GAMP
s’inscrit tout au long de la vie d’un projet. Le
principe fondateur du « Cycle en V ».
3. Les principales différences entre la ver-sion 4 et la version 5GAMP 4 = 5 catégories ; GAMP 5 = 4 catégories.
L’importance des tests concepteurs et four-
nisseurs.
La différenciation entre Qualification et Vali-
dation.
4. Le rôle des utilisateurs tel que défini dans le GAMPDéfinition des rôles et responsabilités des
utilisateurs des systèmes automatisés et in-
formatisés, impliqués dans une démarche de
validation GAMP.
Quels templates utiliser, du Plan Directeur de
Validation aux Rapports de Validation ?
Par quoi commencer une approche de valida-
tion à l’échelle d’un site, d’une division, d’un
groupe, … ?
Comment décliner l’approche globale, sur les
différents systèmes concernés ?
Comment adapter sa stratégie de validation
au contexte système, projet, et opérationnel.
Quels sont les outils pour une maintenance
opérationnelle efficace (SLA, DRP, Change
control, Gestion de configuration, validations
périodiques, …).
Les interactions des utilisateurs avec les four-
nisseurs.
5. Le rôle des fournisseurs tel que défini dans le GAMP Définition des rôles et responsabilités des
fournisseurs de systèmes automatisés et
informatisés, impliqués dans une démarche
de validation GAMP.
Quels templates utiliser, du plan projet aux
fiches de tests ?
Comment rationnaliser la documentation et
l’effort de documentation ?
La version 5 du GAMP apporte un regard nouveau sur les relations utilisateurs / fournisseurs et sur les qualifications. Les notions d’analyses de risques et surtout de gestion des risques sont encore plus présentes. Renforçant sa présence en tant que référence dans de nombreux textes et guides réglementaires du domaine de la santé tant en Europe qu’aux USA, ce nouveau GAMP 5 doit être pris en compte dans les futures stratégies de validation et de qualification des systèmes qu’ils soient informatisés ou automatisés. La version 3 du guide PIC/S PI 011, l’ICH Q9, la future nouvelle version de l’annexe 11 des BPF et l’ASTM E2500 sont aussi à prendre en compte car ils sont en grande partie à l’origine des nouvelles exigences du GAMP 5.
Le rôle de la gestion de projet.
Les interactions des fournisseurs avec les uti-
lisateurs.
6. Résumé du séquençage et les apports de la validationParallèle entre Validation et Développement.
La conformité
La réduction des coûts
La connaissance du système
EXERCICES DE MISE EN SITUATIONPlusieurs exercices sont proposés pendant la
formation pour illustrer les points abordés.
Un QCM vous est proposé afin d’évaluer les
connaissances acquises.
GAMP 5 : Utilisation pragmatique dans la mise en conformité réglementaire
Objectifs
● Comprendre le GAMP, son contexte historique, ses objectifs, son statut de reconnaissance par les instances régle-mentaires.
● Connaître et comprendre les relations entre le GAMP 5 et les autres guides et/ou textes règlementaires.
● Connaître les différences entre GAMP 4 et GAMP 5 et leurs implications sur les validations.
● Savoir utiliser le GAMP 5, son organisation technique, son contenu (méthodologies et templates).
● Comprendre les différences et les points communs avec un schéma de validation traditionnel « Cycle en V ».
● Savoir utiliser les recommandations du GAMP 5 de façon pragmatique.
● Savoir s’appuyer sur les recommandations du GAMP 5 pour se mettre en conformité réglementaire.
● Distinguer les tests de recette/FAT/SAT des tests de validation et savoir intégrer les tests fournisseur dans la validation (Validation Intégrée).
Programme
B34
Formation inter-entreprises
NOUVEAU
DUREE 2 jours
PUBLIC
- Responsable qualité,
- Responsable validation/qualification
- Equipes d’ingénierie
- Equipes de maintenance/métrologie
- Equipes de qualification
- Fournisseurs des industries de la santé
A10
Pour développer vos compétences, nous vous conseillons également :
A11
B9 A17
A18

56
ING
EN
IER
IE /
PR
OJE
TS
Pour tout projet de formation : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
Chaque année, les entreprises ont de nombreux challenges à relever : adaptation à des contraintes légales nationales, européennes ou internationales, nouveaux concepts (commissioning, gestion transverse, …), lancement de nouveaux services ou produits inno-vants, intégration de nouvelles technologies (ERP, LIMS, outils décisionnels,...) ou mise à jour de technologies déjà implantées pour rester compétitif. Si le management de projet est un élément clé de la réussite des projets, la gestion quotidienne est un support indispensable. La gestion de projet a pour vocation de relever ces défis en mettant en place une organisation et une planification de l’ensemble des activités visant à assurer l’atteinte des objectifs du projet (qualité, coûts, délais), effectuer leur suivi, anticiper les changements à mettre en œuvre et les risques, décider et communiquer.
1. Concepts générauxQu’est-ce qu’un projet ?
Terminologie.
Typologie des projets.
Cycles de vie d’un projet.
Exercice.
2. Définition et cadrage du projetConnaître les objectifs du projet et son péri-
mètre.
Définir l’équipe projet : identifier les rôles et
les acteurs décisionnaires.
Définir les besoins : aider le client à remonter
ses vraies attentes.
Rédiger le cahier des charges fonctionnel
(CDCF).
Mise en situation.
Définir les phases et les jalons clés (points
formels de validation).
Définir les taches à réaliser et leur organisa-
tion-: Durée, contraintes, livrables, ressources.
Décomposer le produit du projet (PBS).
Décomposer les taches du projet (WBS).
Estimer le budget (coût et ressources).
Rédiger le cahier des charges du projet
(CDCP) pour contractualiser la réalisation du
projet avec le client.
Mise en situation.
3. Mise en place du processus de contrôleDéfinition du contrôle de l’avancement du projetDéfinir les moyens de contrôle (qualités, délai,
coûts).
Définir le système d’information.
Identifier les différents types de réunions
(lancement, avancement, revue de projet,
comité de pilotage, clôture).
Mettre en place des rapports personnalisés.
Mettre en place un tableau de bord synthétique
et visuel pour informer et rassurer les décideurs.
Etude de cas.
4. Elaboration du planning initial du projetMéthodologie d’élaboration d’un planning.
Planifier les tâches (réseau PERT et diagramme
de GANTT).
Identifier les marges et visualiser le chemin
critique.
Affecter les ressources et analyser les histo-
grammes de charge.
Etablir un plan de charge des ressources pour
obtenir un planning prévisionnel réalisable.
Etablir le planning de référence et positionner
les jalons (décision, milestone).
Exercices.
5. Pilotage du projetRecueillir l’état d’avancement et le reste à
engager.
Calculer l’avancement du projet.
Elaborer les courbes en S.
Re-planifier et gérer les écarts.
Préparer la prise de décision.
Clôturer le projet.
Etude de cas et exercice.
EXERCICES DE MISE EN SITUATION
Plusieurs exercices sont proposés pendant la
formation pour illustrer les points abordés.
Un QCM vous est proposé afin d’évaluer les
connaissances acquises.
Gestion de projet
Objectifs
● Préparer l’ensemble des acteurs d’une
entreprise au travail en mode projet.
● Savoir mettre en place une structure
pour formaliser, planifier, superviser,
contrôler et coordonner les différents
acteurs.
● Connaître les méthodes et outils propres
à la gestion de projet.
● Savoir mobiliser et motiver les acteurs du
projet.
● Savoir planifier et piloter les perfor-
mances, les délais et les coûts d’un projet.
● Savoir mettre en place une communication.
Programme
Pour développer vos compétences, nous vous conseillons également :
C11
C10
Formation inter-entreprises
FORMATION ACTUALISEE
DUREE 2 jours
PUBLIC
- Chefs de projets
- Coordinateurs de projets
- Ingénieurs de projets
- Maîtres d’œuvre, Maîtres d’ouvrage
- Responsables de services et collabora-
teurs impliqués dans un projet
- Toute personne impliquée dans les
projets de l’entreprise
Toutes nos formations sont
disponibles en anglais
Contactez-nous + 212 (0)6 61 13 69 28
www.2ssante.com

ING
EN
IER
IE /
PR
OJE
TS
57Pour toute formation intra-entreprise : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
1. Concepts généraux - Culture de l’entreprise face aux projets.
- Concepts généraux autour du manage-
ment de projet.
- Les différentes approches pour le manage-
ment de projet.
- Les projets et l’entreprise.
2. Evaluer le projet - Etudier les scénarios possibles et choisir un
scénario.
- Analyser et traiter les risques : les menaces et
les opportunités, évaluer les risques majeurs
- Evaluer les objectifs.
- Négocier et vendre son projet.
- Formaliser et communiquer les objectifs du
projet.
Etude de cas.
3. Organiser le projet - Rôle et responsabilités du chef de projet.
- Composer et manager l’équipe projet :
identifier les conditions de l’efficacité d’une
équipe.
- Définir les responsabilités (OBS), les compé-
tences et les ressources (RBS).
- Contractualiser les relations intervenants/
chefs de services/chef de projet.
- Obtenir l’engagement de l’équipe projet.
- Définir les instances de décision.
- Identifier les différents types de réunion
(lancement, avancement, revue de projet,
comité de pilotage, clôture).
- Impliquer toutes les parties prenantes.
- Manager l’élaboration du planning.
- Formaliser un plan de management de projet
(PMP).
Etude de cas.
4. Réaliser le projet - Gérer un moment clé : la réunion de l’avan-
cement.
- Organiser et mener à bien les réunions de
pilotage et revues de projet.
- Anticiper et gérer les aléas et modifications
- Maîtriser les performances, les modifications,
les risques, les délais et les coûts.
- Maîtriser la qualité du projet : assurer la
traçabilité des besoins, traiter les modifica-
tions, faire la revue de projet.
- Faire prendre les décisions.
- Communiquer périodiquement avec un
tableau de bord percutant.
- Détecter et traiter les situations conflic-
tuelles.
- Réagir face à une situation difficile.
- S’affirmer dans les situations relationnelles
difficiles.
Etude de cas.
5. Clôturer le projet- Réceptionner les prestations / installations.
- Transférer l’ouvrage à l’exploitant.
- Clôturer le projet.
Etude de cas.
Un QCM vous est proposé afin d’évaluer les
connaissances acquises.
L’environnement des entreprises est de plus en plus évolutif et la capacité des organisations à réussir au plus vite ses projets stratégiques est une composante essentielle de leur succès. Il faut accorder une attention de plus en plus grande à l’optimisation du processus de réalisation des projets. Le Management de Projet intègre les tâches de Gestion de Projet avec une dimension supplémentaire : la définition des objectifs stratégiques et politiques de la direction (Direction de Projet). Des équipes mobilisées, une hiérarchie judicieusement sollicitée, et des risques et contraintes anticipés vont contribuer à concevoir et réaliser avec succès les projets de l’entreprise.
Management / Direction de projet
Objectifs
● Comprendre la culture du travail en
mode projet.
● Savoir négocier, formaliser et faire valider
les objectifs et le périmètre du projet.
● Savoir organiser le projet autour des
différents acteurs et manager l’équipe
projet.
● Savoir conduire le changement et
s’adapter au type de projet.
● Savoir mettre en place une gestion globale
des risques.
Programme
C11FORMATION ACTUALISEE
Formation inter-entreprises
DUREE 2 jours
PUBLIC
- Chefs de projets
- Coordinateurs de projets
- Ingénieurs de projets
- Maîtres d’œuvre, Maîtres d’ouvrage
- Responsables de services et collabora-
teurs impliqués dans un projet
- Toute personne impliquée dans les
projets de l’entreprise
C10
Pour développer vos compétences, nous vous conseillons également :
Toutes nos formations sont
réalisables en intra-entreprise
Contactez-nous + 212 (0)6 61 13 69 28
www.2ssante.com

58
ING
EN
IER
IE /
PR
OJE
TS
Pour tout projet de formation : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
1. Réglementations et DéfinitionsPrésentation des principales réglementations
et normes dans le domaine de l’ingénierie des
besoins.
L’importance de suivre les bonnes pratiques
d’ingénierie.
Revue des définitions des principaux termes
utilisés dans l’expression des besoins et
les cahiers des charges : Besoin, Exigence,
Contrainte, Cahier des Charges, Spécifications,
Critères d’acceptation, Utilisateur, …
2. Cahier des charges et cycle de vie projetLes différents cycles de vie projet.
Le cahier des charges, un outil de communication
Situer le cahier dans le cycle de vie d’un projet
Notions de Pourquoi, Quoi et Comment.
Ce que le cahier des charges ne décrit pas.
Responsabilités : Qui rédige, vérifie et
approuve ?
Cahier des charges et spécifications.
Cahier et des charges versus Spécifications
des Besoins Utilisateur.
3. Méthodologie d’élaboration d’un cahier des chargesPourquoi a-t-on besoin de rédiger un cahier
des charges ?
Présentation d’une méthodologie d’élabora-
tion d’un cahier des charges en 6 étapes.
4. Constitution du groupe de travailDe qui a-t-on besoin pour rédiger un cahier
des charges ? Utilisateurs « directs » et uti-
lisateurs « indirects ». Qui doit participer à
l’élaboration d’un cahier des charges par
rapport à ce qui doit être pris en compte ?
5. Recueil des besoinsPourquoi les spécifications et le cahier des
charges sont-ils si importants ?
Pourquoi une mauvaise caractéristique n’est
pas un défaut mais une erreur ?
Comment se poser les bonnes questions ?
Comment rendre les objectifs exploitables ?
Comment utiliser le concept « SMART » : Spé-
cifique, Mesurable, Accessible, Réaliste, Tem-
porel ?
Présentation de plusieurs méthodes de recueil
du besoin : Étude marketing, Étude sociolo-
gique, Conception participative, Méthode
qualité, Prototypage, Modélisation des flux.
Typologie des exigences.
6. Consolidation des besoinsHiérarchiser, épurer et prioriser les besoins.
Présentation des critères à satisfaire pour les be-
soins : Cohérence, Abstraction, Non ambiguïté,
La cohérence et la complétude des besoins recueillis auprès des utilisateurs, permettent la rédaction du cahier des charges qui servira d’élément fondamental à la validation des systèmes au regard de la réglementation. Des besoins correctement et complètement exprimés permettent aussi de s’assurer que le système correspondra aux attentes de tous les utilisateurs et sera conforme aux exigences de l’entreprise.
Précision, Lisibilité, Vérifiabilité, Maintenabi-
lité, Traçabilité.
7. Rédaction du cahier des chargesPrésentation d’un modèle de cahier des
charges basé sur l’IEEE 830.
Correspondance Etapes / Document.
8. Revue du cahier des charges Processus de revue et notion d’indépendance
Comment construire une grille de revue ?
Les 3 axes de revue d’un cahier des charges :
Métier, Réglementaire, Qualité.
Comment gérer l’évolution du cahier des
charges ?
9. Application sur un cas concretCréation d’un cahier des charges.
Synthèse de l’exercice : Les difficultés rencon-
trées ; Ce qui semble bien fonctionner ; Ce qui
semble manquer.
EXERCICES DE MISE EN SITUATION
Plusieurs exercices sont proposés pendant la
formation pour illustrer les points abordés.
Un QCM vous est proposé afin d’évaluer les
connaissances acquises.
Analyse des besoins et Cahier des Charges : application à l’informatique ou aux équipements
Objectifs
● Comprendre le rôle clé du cahier des charges dans un projet et dans une va-lidation.
● Savoir situer le cahier des charges dans le cycle de vie d’un projet.
● Connaître la différence entre expression des besoins et Cahier des Clauses Tech-niques Particulières.
● Acquérir une méthodologie pragma-tique pour l’élaboration d’un cahier des charges.
● Connaître les différences entre les besoins, les exigences et les contraintes.
● Connaître les différentes méthodes de recueil des besoins et des exigences.
● Savoir être exhaustif dans le recueil des besoins (besoins implicites et explicites, exigences réglementaires, prise en compte de tous les intervenants, …).
● Savoir rédiger un cahier des charges sans ambiguïté, cohérent, complet, vérifiable.
● Savoir rédiger et vérifier des exigences.
● Savoir constituer une équipe pour recueillir les besoins.
● Savoir prendre en compte et traiter l’évolution des besoins.
● Savoir mettre en place des documents maintenables, « validables » répondant aux exigences réglementaires et nor-matives européennes, américaines ou internationales.
Programme
C20
PUBLIC
Toute personne qui a des cahiers de
charges à rédiger, à vérifier ou à utiliser.
B34
Pour développer vos compétences, nous vous conseillons également :
A17

ING
EN
IER
IE /
PR
OJE
TS
59Pour toute formation intra-entreprise : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
Coût, Qualité, Délai sont trois composantes indissociables pour de nombreux processus, y compris les processus de développe-ment de systèmes informatisés. Afin de maîtriser ce triplet, conduire des tests selon une méthodologie définie et organisée est indispensable. Sans test, le système livré est truffé de bugs, ce qui dégrade sa qualité et impose un grand nombre d’allers-retours entre les utilisateurs et les développeurs, ce qui entraîne des délais, tout en augmentant les couts. CQFD ! L’amélioration du rapport coût/bénéfice d’un système informatique passe par une meilleure pertinence des tests réalisés. L’état de l’art permet aujourd’hui d’aborder ce sujet avec de meilleures armes (génération automatique de pré-requis, scénarii de tests automatisés, …).
1. Pourquoi tester ? Quels sont les enjeux ?
2. Définir une stratégie de tests
3. Préparation des plans de tests, cahiers de recette et autres protocoles
4. La typologie de tests et les différentes techniques de test
5. Organiser l’exécution des tests
6. Les activités de support pour l’exécution des tests
7. La réception des évolutions
8. Les outils d’automatisation
Un QCM vous est proposé afin d’évaluer les
connaissances acquises.
Management des techniquesde tests informatique et recette de logiciel
Objectifs
● Comprendre la nécessité d’une stratégie
de tests formalisée de vos systèmes
d’information.
● Maîtriser les différentes techniques et la
terminologie des tests.
● Rédiger un cahier de recettes, le mettre en
place en évaluant la couverture et l’avan-
cement des tests.
Programme
C22
DES FORMATIONS ADAPTÉES A VOTRE BESOIN
Les formations inter-entreprisesLes formations inter-entreprises sont organisées dans notre Centre de Formation à Lyon. Elles permettent de confronter
vos expériences avec des professionnels de différentes entreprises.
Les formations intra-entrepriseLes formations intra-entreprise sont des formations organisées sur votre site. Elles vous permettent d’assurer la diffusion
d’un message commun à vos collaborateurs et de créer une dynamique de groupe. Toutes nos formations peuvent être
réalisées en intra et adaptées à vos besoins et contextes spécifiques.
Les formations-actionLes formations-action sont des formations terrain réalisées sur votre site. Elles sont construites sur des cas spécifiques à
votre entreprise selon vos objectifs et animées par un formateur spécialiste du domaine.
PUBLIC
Toute personne qui doit réaliser des tests
informatiques.
A11
Pour développer vos compétences, nous vous conseillons également :

60
ING
EN
IER
IE /
PR
OJE
TS
Pour tout projet de formation : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
1. Notions de base en informatiqueHistorique.
Enjeux de l’informatisation.
Définitions et présentation illustrée des termes
couramment utilisés : internet/intranet, client-
serveur, réseau, mémoire, sécurité logique,
sauvegarde, archive,...
2. Organisation des services informatiques en entrepriseL’infrastructure informatique dans l’entreprise
Les fonctions indispensables pour en assurer
la bonne marche : études, maintenance,
production, système, bureautique, réseau.
3. Introduction au génie logicielLes étapes essentielles pour définir, concevoir
et réaliser un logiciel.
L’informatisation d’un processus.
Le cycle de vie d’un système informatique.
L’organisation d’un projet de développement
informatique et les risques associés.
4. Maitrise des risques informatiquesLes risques liés au développement et à
l’exploitation de systèmes informatiques :
l’organisation, les projets, les risques fonc-
tionnels, la sécurité physique et logique, les
sauvegardes, l’archivage, l’environnement,...
Etudes de cas.
Méthodologies pour obtenir un niveau de
confiance acceptable.
Référentiels sur la sécurité informatique.
5. Système qualité informatiqueLes exigences réglementaires.
Les références normatives.
Mise en place d’un système qualité informa-
tique.
6. L’informatique aujourd’huiLes perspectives d’évolution de l’informatique-:
internet, e-commerce, signature électronique,
gestion électronique des documents.
7. Composants informatiquesLes principaux composants d’un système
informatique : unité centrale, carte mère,
connecteurs, rom, bios, horloges internes, chip-
set, processeurs, mémoires, cartes d’extensions.
Les différents types et modes de fonctionne-
ment des supports de stockage de données
numériques.
8. Introduction aux réseaux informatiquesLe fonctionnement des principaux types de
réseaux informatiques.
Cas pratique :
les différents critères déterminant le choix
d’une architecture réseau.
9. Architecture logicielleLes logiciels présents dans un système infor-
matique (systèmes d’exploitation, bases de
données, progiciels…) et leurs interactions.
Etudes de cas : avantages et inconvénients
des différentes architectures logicielles.
Présentation de la notion d’architecture
«client-serveur».
Un QCM vous est proposé afin d’évaluer les
connaissances acquises.
Les systèmes informatisés sont présents pratiquement partout dans les industries de la santé. Ils sont aussi de plus en plus complexes et les comprendre devient indispensable pour toutes les personnes qui doivent définir ou qualifier / valider des systèmes d’information ou des processus de gestion de données ou de documents. Il est donc essentiel, par une approche pragmatique, de savoir en identifier leurs composants et d’en comprendre le fonctionnement. Ceci dans le but de focaliser les efforts de test, de gestion et de contrôle sur les points essentiels les concernant et d’être capable de justifier les stratégies et méthodologies de validation mises en oeuvre.
L’informatique Initiation pour les non spécialistes
Objectifs
● Comprendre les enjeux de l’informatisa-
tion de l’entreprise.
● Comprendre l’organisation des services
informatiques et le rôle des différentes
entités.
● Maîtriser le vocabulaire informatique.
● Découvrir les grands principes du déve-
loppement d’un logiciel.
● Comprendre les risques liés aux dévelop-
pements et à l’exploitation informatique.
● Optimiser ses relations avec les services
informatiques (interne, externe).
● Connaître les composants matériels et
logiciels d’un système informatique :
fonction, criticité, intérêt, rôle dans le
fonctionnement d’un système.
Programme
D12
Formation inter-entreprises
NOUVEAU
PÉRIMÈTRE
• Systèmes informatisés : ERP, LIMS, GED, MES, …
• Infrastructure Informatique
• Réseaux : LAN, WAN, SAN, …
DUREE 2 jours
PUBLIC
- Assurance Qualité
- Production
- Qualification / Validation
- Ingénierie / Maintenance
B14
D13
Pour développer vos compétences, nous vous conseillons également :
A11 A40
B9
Vous souhaitez obtenir
plus d’informations ?
Contactez-nous + 212 (0)6 61 13 69 28
www.2ssante.com

ING
EN
IER
IE /
PR
OJE
TS
61Pour toute formation intra-entreprise : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
1. Historique et définitionsHistorique : pourquoi les systèmes automatisés sont devenus indispensables dans les indus-tries de la santé.Définitions : connaître et comprendre les différents termes qui caractérisent les auto-matismes.
2. Présentation et constitution des systèmes automatisésLes différents types d’architectures : les sys-tèmes de contrôle/commande ouverts, les systèmes de contrôle/commande distribués, notion d’entrées/sorties directes ou déportées.
Les différents éléments matériels constituant un système automatisé: PC, serveurs, auto-mates, réseaux, bus de terrain, capteurs, ac-tionneurs.
Les différents éléments matériels constituant un automate programmable industriel.
Les différents éléments logiciels dans les pc et serveurs : système d’exploitation, logiciels de communication, progiciels de contrôle/commande ou de supervision, applicatifs développés.Les différents éléments logiciels de l’automate : système d’exploitation interne à l’automate, pro-giciel de programmation et types de langage de programmation automate (ladder logic), logiciel de communication, ...
3. Fonctionnement d’un système automa-tiséPrésentation et description des principes d’acquisition d’informations provenant des capteurs, via les entrées du système automa-tisé, ainsi que les transformations permettant à partir d’une information numérique, le pilo-tage des actionneurs via les sorties du système automatisé.Savoir comment un programme est exécuté à
l’intérieur d’un automate programmable. Pré-sentation et description des types de contrôles programmés dans les automates : automatique continue (régulations), automatique logique, automatique séquentielle (grafcet).Les différents principes de transmissions des in-formations entre les automates et les différents PC du système automatisé.Savoir comment interagissent les différents éléments constituant l’interface homme-machine, constituant le moyen de supervi-sion du système automatisé.Comprendre comment les données gérées et générées dans les PC de contrôle/commande peuvent être interfacées avec des systèmes informatiques classiques (bases de données, …). Savoir identifier les points clés essentiels permettant un fonctionnement correct.
4. Methodologie de conception d’un système automatiséConnaître les différentes étapes de dévelo-ppement d’un système automatisé, les données et documents nécessaires à sa conception, les différents métiers et interlocuteurs.Description du cycle de vie d’un système automatisé dans une démarche classique et selon le Gamp 5.Les différents modèles de contrôle-commande industriels : cim, isa. Nécessité d’une approche méthodique de conception : description de la méthodologie préconisée par la norme isa s88 / iec 61512.
5. Les risques lies aux systèmes automatisés
Savoir identifier les et comprendre les risques spécifiques aux systèmes automatisés : - Les risques liés aux différents choix techno-logiques dès la conception
- Les risques liés au choix des fournisseurs - Les risques liées à la l’organisation des responsabilités humaines de gestion du
Les systèmes automatisés sont de plus en plus répandus et omniprésents dans les industries de la santé. Parce qu’ils sont les éléments pilotant et animant tous procédés de fabrication et procédés d’utilité associés, ils sont directement exposés aux inspections. Il est donc essentiel, par une approche pragmatique, de savoir en identifier leurs composants et d’en comprendre le fonctionnement. Ceci dans le but de focaliser les efforts de test, de gestion et de contrôle sur les points essentiels les concernant.
système automatisé - Les risques liés à la documentation - Les risques liés au suivi, à la maintenance et aux modifications apportées
6. La qualification d’un système automatisé
Savoir identifier, organiser et planifier les diffé-rentes étapes de tests et de qualification.Pourquoi il faut séquencer les tests ?Les principaux pré-requis de tests d’un système automatisé. Comprendre les points techniques critiques sur lesquels doivent se focaliser le plus les efforts de tests. Maintien de l’état de validité d’un système automatisé.
L’automatiqueInitiation pour les non spécialistes
Objectifs
● Mieux connaître les spécificités de l’auto-matique industrielle pour les intégrer lors de la mise en place ou de la modification d’unités de production.
● Comprendre les éléments clefs pour dia-loguer avec les fournisseurs internes ou externes de systèmes automatisés.
● Saisir les enjeux liés aux systèmes auto-matisés soumis à réglementation phar-maceutique ou apparentée.
● Situer le rôle des intervenants dans le développement, l’exploitation et la main-tenance des systèmes automatisés (au-tomaticien, métrologue, instrumentiste, etc…).
Programme
D13
Formation inter-entreprises
NOUVEAU
PÉRIMÈTRE
• Systèmes automatisés de production, de gestion des utilités, de gestion de bâtiment, de pilotage d’équipement ;
• Automates, logiciels de supervision, DCS, GTC, BMS … PC et serveurs associés ;
• Programmes automates : logique, régulation, GRAFCET ;
• Réseaux et bus de terrain.
• Norme ISA-S88 / IEC 61512
DUREE 2 jours
PUBLIC
- Assurance Qualité
- Production
- Qualification / Validation
- Ingénierie / Maintenance
D12
Pour développer vos compétences, nous vous conseillons également :
A10
B34
B9
A10

62
ING
EN
IER
IE /
PR
OJE
TS
Pour tout projet de formation : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
Avec un volume de données de plus en plus important à gérer, et des exigences qualités qui s’intensifient, l’intégration d’un LIMS avec ses nombreuses fonctionnalités et ses potentialités par rapport aux besoins d’interfaçage au sein d’une organisation, répond à un enjeu clé de la gestion des résultats au Laboratoire. En effet son domaine d’application ne se limite pas aux strictes prérogatives du Laboratoire, mais s’étend à la gestion des contrôles qualité dans tous les lieux de l’Entreprise ainsi que dans les lignes de fabrication où il s’interface avec les logiciels de GPAO.
1. Environnement actuel du Laboratoire et ses enjeuxLa qualité dans les laboratoires. La composi-tion des outils de gestion de la Qualité. L’évolu-tion des outils de traitement de l’information.
2. Pourquoi un système LIMS ?Définition du LIMS et de la terminologie employée. Les activités de laboratoires infor-matisables avec un LIMS : Analyses, Contrôle Qualité, Recherche & Développement, …. Les acteurs concernés par l’utilisation du LIMS. Les enjeux de l’informatisation avec un LIMS. Les limites d’utilisation. Etude de cas.
3. Création et Gestion des Données Tech-niquesGestion des spécifications. Utilisation de la table d’échantillonnage (fréquences de prélèvement et de test). Gestion des périodes probatoires fournisseurs. Gestion du re-échantillonnage. Gestion des utilisateurs, des équipements de laboratoires, des réactifs, des étalons. Gestion des certifications des techni-ciens. Gestion du flux des analyses.
4. Génération et Gestion des Données DynamiquesFonctionnalités du système dans son utilisa-tion quotidienne en Contrôle Qualité et en Recherche et Développement : - Création du prélèvement, gestion de l’étiquetage et du re-étiquetage des échantillons
- Réception et répartition des échantillons - Attribution analyse / analyste - Calculs, saisies, vérification des données brutes et calculées
- Gestion des certificats d’analyse - Gestion de la certification des techniciens - Sécurité des données - Traçabilité complète des modifications
Planification des analyses. Analyses de ten-dances. Gestion des non-conformités, des
re-tests, de la destruction des échantillons, de l’échantillothèque, des études de stabilité. Gestion des anomalies. Gestion des procé-dures, contrôle d’environnement, métrologie, …. Monitoring des activités et rapports.
5. Architecture des différents LIMS et les systèmes interfacésLes différentes configurations (WEB, ..) Les options d’interfaçage avec les équipements et autres logiciels : GPAO/ERP; systèmes de gestion des données cliniques, outils de repor-ting ; systèmes / équipements de laboratoire : HPLC, CPG, MS, séquenceur; automates : dilu-teurs ; cahiers de laboratoire électroniques, …
6. Intégrer les exigences réglementaires à son LIMSLes référentiels réglementaires applicables en CQ et en R&D. - BPF (France / Europe) et cGMP (FDA) pour le Contrôle Qualité,
- BPL / GLP pour la R&D pré-clinique (Europe, FDA, OCDE), fascicule 10
- BPC / GCP pour la R&D clinique (Europe, FDA, ICH),
- 21 CFR 11 Enregistrements et Signatures Electroniques annexe 11
- PIC/S PI011-3
7. Etat de l’art des solutions disponibles / Le marché du LIMSLes offres du marché. Les solutions et straté-gies des éditeurs. Les solutions techniques.
8. Conduite d’un projet LIMSLa conduite du changement. Promouvoir et lancer le projet : les acteurs du projet, le bud-get, le ROI. La phase de sélection : les besoins utilisateurs, le cahier des charges, le dossier d’appel d’offre, le choix du fournisseur. La gestion de projet : les grandes étapes du projet, ressources et organisation, les clés du succès, les risques, le planning. Les étapes du
projet en détail : les spécifications fonction-nelles, le dossier de paramétrage, le dossier de réalisation, les procédures d’exploitation et d’utilisation, la stratégie d’implémenta-tion. Contextes particuliers : un changement de version, la reprise des données existantes, une migration, …. Etude de cas.
9. Validation du LIMSEnjeux de la validation. Analyse de risque, Démarche de validation : QF, QD, QI, QO, QP. Indépendance entre le cycle de vie d’implé-mentation et les étapes de validation. Les exigences réglementaires liées à la mise en exploitation et à la gestion des modifications. Problématique de la migration des données. Points clés à valider sur un LIMS. Etude de cas.
10. Après le démarrageLa maintenance du système. La gestion des changements.
EXERCICES DE MISE EN SITUATIONPlusieurs exercices sont proposés pendant la formation pour illustrer les points abordés.
Un QCM vous est proposé afin d’évaluer les connaissances acquises.
*Laboratory Information Management System.
Objectifs
● Comprendre la démarche de mise en œuvre d’un LIMS, depuis l’expression des besoins, le cahier des charges, la sélection jusqu’à la mise en exploitation et sa vali-dation.
● Savoir définir les fonctionnalités d’un LIMS tant en Contrôle Qualité qu’en Recherche et Développement.
● Connaître les exigences réglementaires applicables pour les intégrer au LIMS-: (BPF cGMP, BPL/GLP, BPC/GCP, ICH, 21 CFR 11 Enregistrements et Signatures Electroniques, annexe 11 des GMP, PIC/S PI011-3).
● Définir des outils de pilotage du projet et
les facteurs clé du succès.
Programme
LIMS* : fonctionnalités, conduite du projet et validation
D15
PUBLIC
- Chef de projet utilisateur
- Chef de projet informatique
- Key user / utilisateur référent
- Responsable qualité
- Equipe d’ingénierie
- Responsable de laboratoire
B33
D26
C10
A11

ING
EN
IER
IE /
PR
OJE
TS
63Pour toute formation intra-entreprise : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
La maîtrise de l’environnement des équipements de production et de contrôle (conditionnement d’air des salles blanches, hygrométrie, pression…), des fluides utilisés dans les processus de production et de contrôle des produits (eaux, air, …) doit se faire dans le respect des exigences réglementaires et normatives. Toutefois il n’est pas toujours facile de s’y retrouver entre ces textes réglementaires et les normes et de séparer ce qui est indispensable de ce qui est superflu voire contre productif. Cette formation, qui n’a pas pour but de vous apprendre a concevoir l’un de ces systèmes, va vous apporter les bases nécessaires pour écrire ou vérifier un cahier des charges (SBU / URS) ou un dossier de qualification / validation concernant les principales utilités de votre entreprise.
1. Eau et VapeurDéfinitions concernant l’eau et la vapeur.Réglementations et normes concernant l’eau et la vapeur (Europe, USA et International).Principes de conception des systèmes de pro-duction et distribution d’eau et de vapeur :- Résines échangeuses d’ions- Osmose inverse- Distillateurs multi effets- Distillateurs à thermo compression- Générateurs vapeur propre et pure- Etc.…Commissioning, Qualification et Validation des systèmes de production et de distribution d’eau et de vapeur : - Les risques spécifiques aux systèmes d’eau et de vapeur
- Les points critiques à ne pas oublier dans le cahier des charges (SBU / URS)
- Les tests de qualification spécifiques aux systèmes d’eau et de vapeur
2. Air comprimé et Gaz spéciauxDéfinitions concernant l’air comprimé et les gaz spéciaux. Réglementations et normes concernant l’air comprimé et les gaz spéciaux (Europe, USA et International). Conception des systèmes de production de gaz :- Air Comprimé- Azote- Etc. …Commissioning, Qualification et Validation des systèmes de production et de distribution d’air comprimé : - Les risques spécifiques aux systèmes de production et de distribution d’air com-primé
- Les points critiques à ne pas oublier dans le cahier des charges (SBU / URS)
- Les tests de qualification spécifiques aux systèmes de production et de distribution
d’air comprimé - Commissioning, Qualification et Validation des systèmes de production et de distribu-tion des gaz spéciaux :
- Les risques spécifiques aux systèmes de pro-duction et de distribution des gaz spéciaux
- Les points critiques à ne pas oublier dans le cahier des charges (SBU / URS)
- Les tests de qualification spécifiques aux systèmes de production et de distribution des gaz spéciaux
3. Conception des zones à atmosphères contrôlées Définitions concernant les zones à atmos-phères contrôlées.Réglementations et normes concernant les zones à atmosphères contrôlées (Europe, USA et International).Les principes de conditionnement d’air dans les salles :- Température- Hygrométrie- Différence de pression- Flux- ParticulesPrincipes de conception des zones à atmos-phères contrôlées.Les points critiques à ne pas oublier dans le cahier des charges (SBU / URS).Principes de qualification des zones à atmos-phères contrôlées (typologie des cas de tests applicable en QI, QO et QP).Les cartographies des salles.
Un QCM vous est proposé afin d’évaluer les connaissances acquises.
Les Utilités : points-clés pour l’expression des besoins et la validation
Objectifs
● Savoir maîtriser l’environnement de production (HVAC et zone à atmosphère contrôlée).
● Savoir maîtriser les utilités dans les pro-cessus de production et de contrôle (eau, vapeur, air comprimé, azote).
● Connaître les réglementations Euro-péennes, Américaines et internationales applicables aux principales utilités.
● Connaître les points clés concernant les utilités.
● Connaître les risques concernant les utilités.
● Savoir définir les spécifications des utilités (SBU / URS) en contexte réglementaire.
● Savoir qualifier les systèmes de traitement d’air et les utilités.
Programme
D22
Formation inter-entreprises
FORMATION ACTUALISEE
PÉRIMÈTRE
• Zones à Atmosphères Contrôlées (ZAC)
• Génération et distribution d’eau
• Génération et distribution de vapeur d’eau
• Génération et distribution d’air com-primé
• Gaz spéciaux
DUREE 2 jours
PUBLIC
Toute personne qui a besoin d’écrire
ou de vérifier un cahier des charges
concernant les utilités ou qui a besoin
de qualifier ou de vérifier les dossiers de
qualification d’utilités :
- Assurance Qualité
- Production
- Qualification / Validation
A10
Pour développer vos compétences, nous vous conseillons également :
A18 A17
C20

64 Pour tout projet de formation : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
GxP Manager intègre un moteur de liens de nouvelle génération qui vous permet de lier et
gérer vos données techniques et réglementaires. Ainsi, vous disposez d’un seul outil pour
gérer les exigences projets, produire automatiquement des matrices de traçabilité, créer
des workflows, exécuter des tests et publier en temps réel vos documents techniques. Vous
pouvez aussi gérer facilement les accès et authentifier vos documents avec des signatures
électroniques.
Avec sa nouvelle version Entreprise, Validatool change de dimension et devient GxP
Manager ! Leader dans son domaine avec plus de 30 références dans les Industries de la
Santé, GxP Manager est aussi présent aux Etats-Unis.
SOLUTIONS MÉTIERSIngénierie, Informatique, Laboratoires et Validation
GxPMANAGER
LOGICIEL INTÉGRÉ DE GESTION DES DONNÉES
MÉTIERS, PROJETS ET RÉGLEMENTAIRES
• Etat des lieux pharmaceutique
• Plan directeur de validation
• Gestion des bâtiments et des locaux
• Gestion des équipements et des procédés
• Gestion de la réglementation
• Management du risque
• CAPA
• Gestion des exigences et des spécifications
• Analyses de risques (AMDEC, HAZOP, HACCP)
• Protocoles de Qualification (QD, QI, QO, QP)
• Exécution des tests et rapports de validation
• Matrices de traçabilité
• Gestion des modifications
• Planification
GxPMANAGER
Pour tous vos projets : GXP MANAGER
INDUSTRIES DE LA SANTÉ
Téléphone : +33 (0) 426 100 810
Email: [email protected]
www.gxpmanager.com

ING
EN
IER
IE /
PR
OJE
TS
65Pour toute formation intra-entreprise : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
Introduction et notions fondamentales
La reprise de fonds documentaire : Comment passer du papier au numérique ?
- Qualité et volume du fonds documentaire
- Numérisation des documents
- Classement et indexation
- RAD, LAD et OCR
- Les clés de la réussite d’un projet de reprise
de fonds documentaire
- Études de cas
Gestion électronique de documents : Au delà de l’outil, une problématique
d’entreprise.
- L’architecture des systèmes de GED
- Les classements et la hiérarchie documen-
taire
- Le processus de création des documents
- Le cycle de vie des documents
- Le travail collaboratif
- La distribution et la publication de l’infor-
mation
- Les outils du marché : risques et bénéfices
- Les projets de GED face à la complexité des
besoins
- Études de cas
Archivage et sauvegarde des documents : Assurer la conservation des documents.
- Définition de la stratégie
- Supports de stockage
- Organisation du stockage
- Durée de conservation
- Étude de cas
Évolutions et perspectives : Anticiper, évaluer, initier la mutation des
systèmes d’information.
- Déstructuration documentaire
- Transition vers la GEI
- Bases de données et technologies XML
- Les moteurs de recherche « intelligents »
- Les choix stratégiques : processus, besoins,
outils et architecture
- L’accompagnement à la gestion du chan-
gement
Un QCM vous est proposé afin d’évaluer les
connaissances acquises.
L’information qui circule dans l’Entreprise devient de plus en plus complexe. Le document papier a presque disparu, le document électronique perd sa structure et l’information transite de plus en plus sur les Intranet et les messageries d’entreprise. Transférer les archives papier dans des armoires électroniques, mettre en place une solution de GED (Gestion Électronique de Documents), en intégrant tous les supports d’information, permettent aux entreprises de gagner 10 à 20 % d’efficacité dans le travail. C’est donc un enjeu économique majeur dans un domaine fortement concurrentiel. Les tendances actuelles en matière de gestion de l’information, appuyées sur de nouvelles technologies, vont renforcer la pertinence et la fiabilité de l’information et développer le travail collaboratif. L’enjeu est encore plus important, et la maîtrise de l’ensemble de la chaîne documentaire avec la transition vers une GEI (Gestion Electronique de l’Information) est une des clés du succès de la mutation progressive des systèmes d’information de l’entreprise.
De la Gestion Électronique de Documents (GED) à la Gestion Electronique de l’Information (GEI)
Objectifs
● Revoir les notions fondamentales de GED
et des systèmes d’Information.
● Appréhender les projets de reprise de
fonds documentaire.
● Comprendre les fonctions des systèmes
de gestion documentaire.
● Comprendre les enjeux et maîtriser les
risques de la mise en place d’un projet de
GED.
● Connaître les évolutions et les tendances
dans le domaine de l’information.
● Savoir passer d’une GED à une GEI.
Programme
D26NOUVEAU
Formation inter-entreprises
DUREE 2 jours
PUBLIC
- Responsables de projets Business / IT
- Affaires Réglementaires, responsables
d’études pré-cliniques/cliniques
- Responsables des archives
- Responsables de services et collabora-
teurs impliqués dans un projet de GED
- Toute personne impliquée dans les
projets de l’entreprise
A11
Pour développer vos compétences, nous vous conseillons également :
B27
Toutes nos formations sont
disponibles en anglais
Contactez-nous + 212 (0)6 61 13 69 28
www.2ssante.com

MÉTROLOGIE

ME
TR
OLO
GIE
67Pour toute formation intra-entreprise : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
Vérification, étalonnage, plage et incertitude de mesure, reproductibilité, répétabilité, justesse, erreur, … La terminologie de la métrologie est le propre de ce métier et son implication dans l’entreprise primordiale. Comprendre facilement le « jargon » de la métrologie, les rouages de ce service et les interactions avec les autres services est important pour tout acteur au quotidien.
1. Le pourquoi de la métrologieSavoir ce qu’est la métrologie.
Comprendre l’intérêt de la métrologie pour
une entreprise.
2. Métrologie et terminologieConnaître et comprendre les termes qui
caractérisent la métrologie.
Exercice.
3. Métrologie et organisationComprendre la notion de traçabilité : chaîne
d’étalonnage ; de l’entreprise aux Etalons
nationaux, voir internationaux.
Exercice.
4. Le processus Métrologie dans l’entre-priseSchéma de mise en œuvre du processus
métrologie. Les pratiques de base. Le recen-
sement. Etapes-clés qui permettent de définir
le programme métrologique. Les interventions.
Suivi de l’équipement. Etude d’un cas pratique.
5. Métrologie et validationSavoir différencier métrologie / validation.
Être capable d’auditer la partie métrologie
lors de la qualification d’un équipement.
Etude de cas : les balances, les pipettes (est-ce
de la métrologie ou de la qualification ?).
EXERCICES DE MISE EN SITUATIONPlusieurs exercices sont proposés pendant la
formation pour illustrer les points abordés.
Un QCM vous est proposé afin d’évaluer les
connaissances acquises.
La Métrologie : notions de base
Objectifs
● Comprendre le rôle essentiel de la métro-
logie pour une entreprise.
● Connaître la terminologie associée.
● Connaître les activités et l’organisation
d’un service métrologie.
● Différencier la métrologie de la validation
et comprendre leurs relations.
Programme
B13
PUBLIC
- Toute personne travaillant en relation
avec un Service Métrologie
- Assurance Qualité
- Validation
D7
Pour développer vos compétences, nous vous conseillons également :
A10
Vous souhaitez obtenir
plus d’informations ?
Contactez-nous + 212 (0)6 61 13 69 28
www.2ssante.com

68
ME
TR
OLO
GIE
Pour tout projet de formation : +212 (0)6 61 13 69 28 ou [email protected] www.2ssante.com
1. Le pourquoi de la métrologieSavoir ce qu’est la métrologie. Comprendre l’intérêt de la métrologie pour une entreprise.
2. Métrologie et terminologieConnaître et comprendre les termes qui carac-térisent la métrologie. Connaître la notion de capabilité. Connaître et comprendre le terme « incertitude », avec ce qui se cache derrière. Exercice.
3. Métrologie et organisationConnaître la différence entre métrologie légale et métrologie scientifique et tech-nique. Connaître l’organisation de la métrologie légale. Connaître l’organisation de la métrologie scientifique et technique. Comprendre la notion de traçabilité : chaîne d’étalonnage : de l’entreprise aux Etalons nationaux, voir internationaux. Exercice.
4. Métrologie et référentielsConnaître le panorama des différentes exi-gences réglementaires en métrologie dans les référentiels ISO / GMP. Avoir une synthèse des exigences. Connaître les outils d’aide à la mise en œuvre de la métrologie (normes et guides).
5. Le processus métrologie : application à l’entrepriseSchéma de mise en œuvre du processus métrologie. Les pratiques de base. Les procé-dures clés. Les processus supports clés. Etude d’un cas pratique.
6. Le processus métrologie : étude du besoin en mesureConnaître les exigences liées à la métrologie.Traduire ces exigences dans un cahier des
charges. Savoir auprès de qui collecter ces exigences. Exercice.
7. Le processus métrologie : le recensementComprendre l’intérêt du recensement. Connaître le type de données à prendre en compte. Repérer et identifier les équipe-ments.Enregistrer les données dans un outil de ges-tion de parc. Etude d’un cas pratique.
8. Le processus « métrologie » : la défini-tion du programme métrologiqueConnaître quelles sont les étapes clés qui permettent de définir le programme métro-logique. Maîtriser la sous-traitance. Docu-menter les spécifications de contrôle. Docu-menter les méthodes d’étalonnage. S’assurer de la formation des intervenants. Etude d’un cas pratique
9. Le processus « métrologie » : les interven-tionsConnaître les points auxquels doivent être formés les opérateurs. Connaître les règles d’application des modes opératoires. Savoir rédiger un compte rendu de résultats. Connaître le type d’étiquette à apposer sur l’équipement. Informer le client à chaque étape. Pouvoir appréhender le traitement des écarts.
10. Le processus « métrologie » : le suivi équipementConnaître le processus de mise à jour des données métrologiques. Savoir gérer les étalons. Archiver les enregistrements de métrologie à la fin de vie d’un équipement.
Selon les exigences réglementaires et normatives, les appareils ou leurs composants sont soumis à des contrôles afin de garantir les mesures qu’ils fournissent ou les sécurités nécessaires pour lesquels ils sont utilisés. De la vérification à l’étalonnage, de la plage à l’incertitude de mesure, en passant par reproductibilité, répétabilité, justesse, la terminologie propre au métier de métrologue permet d’évaluer les besoins de l’entreprise en terme de mesure et de garantir la justesse des mesures réalisées. En raison des contraintes réglementaires et de production, une organisation adéquate des processus métrologiques est cruciale.
11. Métrologie et validationSavoir différencier métrologie / validation. Être capable d’auditer la partie métrologie lors de la qualification d’un équipement. Etude de cas : les balances, les pipettes (est-ce de la métrologie ou de la qualification).
12. Métrologie et sous-traitance Connaître les différents niveaux de sous-traitance. Appréhender le besoin de sous-traitance. Sous-traiter jusqu’où ? Connaître les exigences vis-à-vis des fournisseurs.
13. Les outils d’aide à la métrologieConnaître les logiciels de gestion d’un parc de mesures. Connaître les logiciels d’aide technique et scientifique à la mesure. Etude d’un cas pratique
EXERCICES DE MISE EN SITUATIONPlusieurs exercices sont proposés pendant la formation pour illustrer les points abordés.
Un QCM vous est proposé afin d’évaluer les connaissances acquises.
Objectifs
● Comprendre le rôle essentiel de la métro-logie pour une entreprise.
● Connaître la terminologie associée et les risques à ne pas utiliser le bon terme.
● Connaître les activités et l’organisation d’un service métrologie dans un cadre national et international.
● Connaître les exigences réglementaires et les référentiels normatifs applicables à la métrologie (ISO 17025).
● Différencier la métrologie de la validation et comprendre leurs relations
● Connaître les logiciels d’aide à la mé-trologie : comprendre leur implication à l’utilisation.
● Être capable d’organiser et de suivre la mise en place du processus métrologie conformément aux textes en vigueur.
● Savoir évaluer et optimiser un processus métrologie.
Programme
La Métrologie d’un point de vueorganisationnel et opérationnel
D7
Formation inter-entreprises
DUREE 2 jours
PUBLIC
- Toute personne acteur dans un Service
Métrologie
- Assurance Qualité
- Responsable Validation
- Responsable Qualification
- Responsable Technique
B13
Pour développer vos compétences, nous vous conseillons également :
A10
B104

© C
yber
Co
nse
il. T
ou
s d
roit
s ré
serv
és. C
yber
Co
nse
il SA
S au
cap
ital
de
5 21
8 00
0 €
- 74
rue
de
Bo
nn
el 6
9003
Lyo
n -
SIR
EN 4
47 9
87 4
21 R
CS
Lyo
n
Renseignements et inscriptions
France - CVO-CyberConseil
+ 33 (0)4 72 84 61 33Fax +33 (0)4 26 10 08 [email protected]
Maroc - 2S Santé
+ 212 (0)6 61 13 69 28Fax +212 (0)5 22 36 79 [email protected]
FORMATIONS
2010Validation / Qualification
Qualité / Réglementation
Gestion des risques
Audits / Inspections
Ingénierie / Projets
MétrologieINDUSTRIES DE LA SANTÉ
CVO-CyberConseilLe partenaire des Industries de la Santé


















